







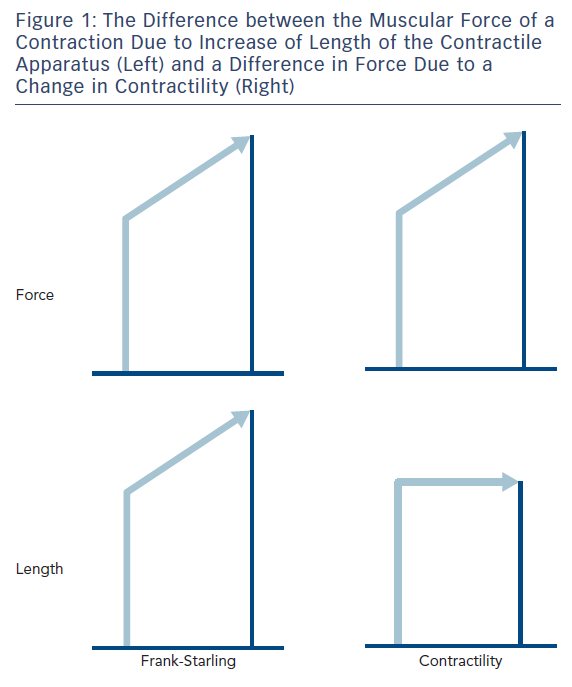
The simplicity of the concept of contractility is illustrated in Figure 1, which is the case for a strip of muscle connected to a force transducer. If one records the force of a contraction, and then measures it again at a longer length, the force is found to be higher. If by making other interventions, such as increasing the calcium ion (Ca2+) concentration of the extracellular fluid, and the force is higher, it is called increased contractility, or lower contractility if the force is lower if the contractility is lower in all the fibres in a heart, could that be called heart failure? Therefore, can you measure contractility in heart failure?
Cardiac Muscle Mechanics
The fundamental mechanism of cardiac muscle was identified in strips of heart muscle from which the cell membrane had been removed in a solution where intracellular Ca2+ was simulated, so the length-force curve could be measured at different Ca2+ concentrations, i.e. complete lengthforce curves at different levels of contractility via Ca2+.1 The purpose of muscle contraction is to create movement, so one also needed to characterise the way cardiac muscle shortened. Early attempts involved the assumption of a model of muscle that had a contractile unit in series with an elastic element, but this was found to be incorrect2 and a complete characterisation had to wait for the development of the laser diffraction technique for measuring sarcomere length (SL).3 Sarcomere shortening velocity has an inverse hyperbolic relationship to force (load). Increasing SL increased velocity at any given load except in the absence of any load (maximum velocity V0). With an increased contractility (increased Ca2+), velocity increased at all levels of load.3
An attempt was made to apply the series elastic model to assessing contractility in patients by measuring Vmax as dP/dt/P, where dP/dt is the rate of rise of left ventricular pressure (LVP) and P is the LVP. Not only was the model wrong, but which P do you take? If one takes the developed pressure above diastolic, Vmax starts at zero and dP/dt/P is infinity, which is absurd. If you take the total pressure, dP/dt/P can be anything you like (according to your zero pressure reference value) and Vmax is not of use.4 A simpler and quite useful index is dP/dtmax, the maximum rate of rise of LVP, which, by chance, is not affected much by end-diastolic volume (EDV) in the intact human, but does reflect changes in contractility.5 However, one can use this method to assess changes in contractility in a given patient, in response to an intervention, but not compare contractility in a patient with heart failure with a normal person because the rate of pressure rise also depends on synchronicity of contraction of the whole left ventricle.
Frank and Starling
Otto Frank (1865–1944)6 and Ernest Henry Starling (1866–1927)7 were contemporary physiologists who both studied the mechanical function of the heart. They both correctly realised that a strip of heart muscle, as with skeletal muscle, developed more force if you increased its length. Starling expressed this as the energy of contraction being dependent on initial length (Starling’s Law),7 but energy is not the same as force. They also both tried to elicit this property in the whole heart, something that is still a difficult exercise today. Starling did not actually perform the necessary experiment to justify his law, because he showed that stroke volume (SV) increased when one increased the EDV. This is a different property that can be demonstrated in the whole heart, which is nothing to do with force depending on length but shows the heart contracts down to the same end-systolic volume (ESV). Frank first showed that isovolumic pressure increased with increase of EDV, and recently I have been involved in helping a German team who are hoping to publish all Frank’s papers in English. It is already clear from these papers that Frank also measured pressure-volume loops, which were later popularised by Sagawa and Suga.8
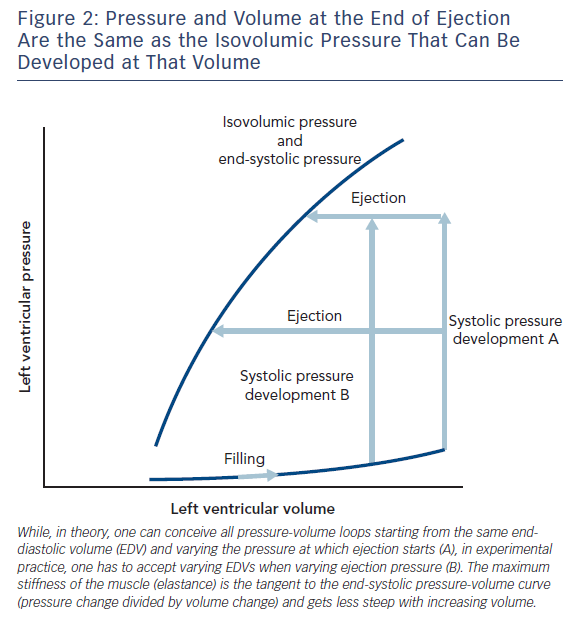
Measurement of peak isovolumic pressure (Frank curve)6 was unknown to Starling and is the nearest one can get, in anaesthetised animal experiments, to the force-length relationship of the isolated muscle strip. When the heart is allowed to fill, develop pressure and then eject a stroke volume, one finds the ejection ends at a pressure the same as obtained isovolumically at the same volume.9
I have omitted from this discussion the many papers of Suga and Sagawa because their laboratory always came out with an end-systolic pressure-volume (ESPV) curve that is straight. The slope of the line is called Emax for maximum elastance. The concept of muscular elastance is correct; it expresses the change in force for a given change in length (for the heart the change in pressure for a given change in volume) and is the reciprocal of compliance. Essentially the stiffness (elastance) of the muscle rises and wanes with the time course of the contraction and is proportional to the force. However, I do not think it is linear, so the elastance is the tangent of the pressure-volume curves, which become less steep as the volume increases (Figure 2). Nevertheless, the tangent at any one volume becomes steeper with time during contraction and less steep with time during relaxation.
Pressure-volume loops can be recorded in patients using a catheter in the left ventricle that transduces pressure conventionally but also carries electrodes to measure capacitance as an index of volume. By varying the venous return with a vena caval balloon one can record a series of pressure-volume loops and plot the ESPV. One can also introduce an intervention that increases contractility and record a leftward shift of ESPV. ESPV is regarded as a more correct index of contractility than LVdP/dtmax (or maximum acceleration of blood from the left ventricle).10 A reduction in contractility could be induced by getting the subject to breathe carbon dioxide,11 but I am not aware that this has been done while measuring human PV loops. This PV loop method was used to confirm changes in contractility from beat to beat during AF. The difficulty is that ejection pressure is virtually clamped by the aortic pressure, and thus I was not prepared to change LVP with a balloon. Nevertheless, results showed ESV varied, i.e. on strong beats the ventricle emptied more and on weak beats it emptied less, indicating a shift of ESPV from beat to beat.12
Ejection Fraction
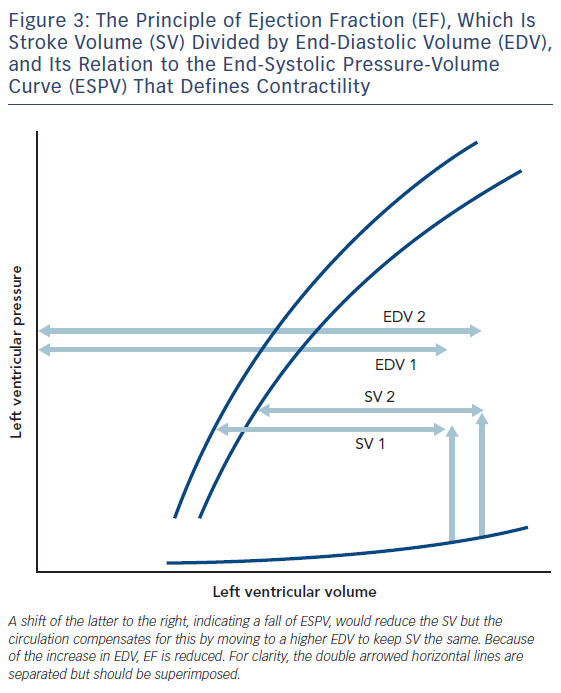
Let us imagine the result of a global fall in contractility of the heart, which might be a way of defining heart failure. In Figure 3, this is depicted as a fall to the right of the ESPV. Assuming that fluid retention, if present, has been removed, the heart compensates to keep cardiac output normal by dilating to a new EDV, and thus a reduction in ejection fraction (EF). According to the British Heart Foundation, heart failure is denoted by an EF less than 55 %. Solaro considers that heart failure is a failure of contractility and states, “Disorders of the heart, whatever the cause, remain epidemic in world heath, and detailed understanding of control of cardiac contractility is essential in the quest for measures to prevent, diagnose, and treat these disorders”.13 There is vast literature testing various theories about the metabolic substrate that underlies reduced contractility in heart failure.
However, the above example of global fall in contractility of the heart is not realistic. I suppose it might apply to some dilated cardiomyopathies, but what about the common causes, such a coronary disease and the end-stage of untreated hypertension; and in heart failure of hypertrophic cardiomyopathy, I can imagine that contractility is increased. After MI, a scar may form and become a sort of series elastic element for the rest of the heart; therefore, the contractility of the scar is obviously nil, but if you look at the contracting muscle, the contractility may be normal or even increased, maybe due to increased sympatho-adrenergic stimulation. Can one regard heart failure, caused by AF, a failure of contractility, when actually, contractility is changing all the time from beat to beat?12
The Confusion of Understanding Heart Failure
A consequence of the adoption of many theories with similar views to Solaro13 is that treatments are devised for heart failure that “manipulate cardiac contractility”.14 A favourite drug to treat heart failure used to be stimulation with dobutamine. The resulting problems actually led to the opposite treatment, namely beta-adrenergic blockade15 or to adrenergic stimulation on top of beta-blockade.
In 2003 the journal Circulation Research published a series of review articles entitled, ‘Unanswered Questions in Heart Failure’. The unanswered questions were:
- Is depressed myocyte contractility centrally involved in heart failure?
- What is the role of beta-adrenergic signalling in heart failure?
- What causes sudden death in heart failure?
- Is abnormal cell growth and hypertrophy the cause of heart failure?
- Does energy starvation cause heart failure?
- What mechanisms underlie diastolic dysfunction in heart failure?
The first of these questions is relevant to the title of the present paper and remained unanswered at the end of the review article.16 The second paper,17 in trying to find some understanding of the apparently illogical success of beta-blockade therapy, got into a tangle of various beta receptor subtypes that did not reveal an answer. The last three questions postulate causes of heart failure other than contractility failure.
What Does Heart Failure Do To Cardiac Cells
One can induce heart failure in dogs by pacing the ventricle at 250 BPM for 6 weeks. It has been claimed that myocytes obtained from such animals were longer and thinner than those from controls, and shortening in response to electrical stimulation was reduced.18 Unfortunately, this cannot be accepted as evidence for reduced contractility in heart failure because: myocytes get damaged in the process of isolation and those from heart-failure animals may be more susceptible to such damage; and shortening is not contractility but load dependent, and the load in these experiments is not known. While in the animal, the load would have been abnormally high due to increased wall force in the dilated heart according to the LaPlace relationship. Therefore, studying isolated myocytes has its limitations.
Therapeutic Implications of the ‘Heart Failure Equals Contractility Failure’ Theory
It was logical to counteract supposed contractility failure with a drug that increases contractility (‘positively inotropic’). Such a drug should improve contraction whether or not there is contractility failure. Adrenergic stimulation also increases heart rate and both effects increase the demand of the heart for oxygen, so these drugs should only be used for testing, e.g. dobutamine during echocardiography if one is looking for poorly contracting segments that might respond. However, one can also increase contractility with digoxin and with the phosphodiesterase 3 inhibitors, of which the best known is milrinone. Digoxin (which Frank found to be positively inotropic)19 is very useful in controlling heart rate in patients with AF and has proponents for its use in patients with heart failure in sinus rhythm, although this is controversial.20
Milrinone has the advantage that it is effective in the presence of betablockade with no effect on heart rate.21 Digoxin and milrinone should probably only be used in acute heart failure as a temporary measure, because they work by increasing intracellular Ca2+. This, in turn, increases the risk of arrhythmias and when given chronically increases mortality, giving milrinone the nickname ‘killrinone’. There are also reports of poor outcomes with digoxin given during sinus rhythm.20 However, improved mortality rate is not everything and in a disease that is fatal, a drug like this might improve the quality of a shorter life.
Another approach is to keep intracellular Ca2+ constant but increase the sensitivity of the contractile proteins to Ca2+, thus predicting a lesser risk of arrhythmias.22 A similar drug produced a benefit in dogs with tachycardia-induced heart failure,23 but I am not aware of its clinical development, possibly because it induced arrhythmias in rat hearts.24
Conclusion
Possibly, heart failure in dilated cardiomyopathies is due to a global reduction in ventricular contractility. In other types of heart failure, the presence or absence of reduced contractility is unknown. In particular, if the failing heart is subjected to coronary disease in some parts but not others, it is very difficult to determine the contractile state of the unaffected parts. In the case of anatomical abnormalities, e.g. valvular disease and congenital heart disease, the heart may be failing globally but myocardial contractility may be normal. Maybe myocardial contractility failure precipitates increased global failure? With the difficulty in measurement, the complicated nature of myocardial fibre orientation and hypertrophy, it is impossible for me to give a firm opinion. Another paper could be written reviewing all that has been written on these aspects, and are still being written.