










With an average incidence of 3.2 per 1,000 person-years, the ominous syndrome of heart failure (HF) affects 1–3% of the general adult population.1 Although the pivotal role of congestion in HF is widely acknowledged, its association with disease progression remains elusive, often resulting in inadequate management strategies.2 Thus, despite significant improvements in mortality rates across virtually all age groups, owing to advancements in both pharmacological and device-based therapies, the prognosis of HF remains unsatisfactory, often trailing behind the outcomes observed in most malignancies.3,4 In fact, acutely decompensated HF stands as one of the leading causes of hospitalisation, with a significant majority of these cases attributed to congestion.5,6 The matter in question could be approached from multiple standpoints.
First, classical pathophysiological explanations oversimplify congestion to salt and water retention in extravascular spaces, although such changes are commonly not the principal mechanism of decompensation.7 As decongestion therapies do not always target every phenotype to the same extent, accurate recognition of phenotype may lead to direct therapeutic benefits. The issue is further complicated by the fact that heterogeneity in congestion phenotypes is a changing landscape, and dissimilar phenotypes might be encountered in the same patient at different time points.8 Additionally, the traditional view of congestion as a surrogate for HF severity understates its causal involvement in HF pathophysiology and organ damage progression.2
Second, the progression of HF and severity of congestion are not in a linear relationship; that is, some patients with severe left ventricle (LV) dysfunction have normal volume status, whereas others can be severely congested without substantial evidence of structural heart abnormalities. Accordingly, clinical signs and symptoms offer limited accuracy in assessing and quantifying congestion, particularly in terms of evaluating therapeutic responses.9,10
Third, although various biomarkers can provide insight on the type and the extent of congestion, their clinical utility is impeded by several factors. As further discussed, a biomarker does not necessarily reflect each congestion phenotype. For instance, natriuretic peptides correlate well with intravascular, whereas carbohydrate antigen 125 (CA125) levels mostly reflect the extent of extravascular congestion.11 However, studies assessing the utility of biomarkers in monitoring the success of decongestion therapy yielded inconclusive results.12,13 Moreover, most blood biomarkers exhibit considerable variability within individuals, and their interpretation in the context of HF is hindered by various comorbidities, such as obesity, AF and chronic kidney disease.14,15
In this review, we aim to summarise contemporary views concerning the pathophysiology of congestion in HF, with particular focus on its association with biomarkers and clinical signs and symptoms, as well as concurrent implications to a tailored approach to congestion in HF patients.
A Multifaceted Nature of Congestion in Heart Failure
In the setting of HF, congestion is defined by signs and symptoms of extracellular fluid accumulation associated with increased cardiac filling pressures.16 However, the term is often wrongly used interchangeably with ‘volume overload’ in daily practice. Despite many patients presenting with decompensated HF having an excessive amount of body water, it has been recently demonstrated that more than half of acute HF patients gain <1 kg in the month preceding the hospital admission.17,18 In contrast, studies based on implantable devices clearly showed an increase in pressures during the same period, thus implicating that the increase cannot be solely attributed to volume expansion.19
The pathophysiological mechanisms underlying the increase in cardiac filling pressures and consequent symptomatic clinical congestion in HF are intertwined within a highly complex dynamic network. The principal underlying factors include impaired systolic and/or diastolic dysfunction, impaired sodium and water balance in the kidneys, sympathetic nervous system (SNS) activation, impaired integrity of the endothelium, and changes in venous capacitance and the properties of the interstitium. Importantly, the relative contribution of each of these processes to the development of decompensated HF is variable, not only between patients, but also within the same patients at different timepoints. Moreover, conforming to the above-noted distinction between volume redistribution and volume overload, underlying mechanisms may contribute unequally to each type of congestion.
SNS overactivity represents a pivotal pathophysiological mechanism operative in HF.20 In fact, cardiac sympathetic output in HF is directly proportional to the increase in pulmonary artery pressure and pulmonary capillary wedge pressure (PCWP).20 SNS overactivity and accompanying parasympathetic withdrawal appear to stem from a maladaptive compensatory response intended to maintain cardiovascular homeostasis, although it is worth mentioning that underlying mechanisms are multifactorial.21 Specifically, increased adrenergic tone can acutely increase cardiac output and redistribute blood to vital organs.21 Nevertheless, even in the acute setting, these effects can prove deleterious, as SNS activation can promote ischaemia by increasing energy expenditure; trigger ventricular tachycardia, especially in the ischaemic setting; promote volume expansion, as further discussed; and cause further increase in filling pressures by venoconstriction.21 The latter mechanism actually emerged as one of the principal culprits behind acute decompensation.7,22
The concept suggests that a multitude of factors, such as myocardial ischaemia and worsening renal function, provoke venoconstriction in splanchnic venous beds by SNS activation. This, in the context of preexisting interstitial volume overload, precipitates rapid redistribution of fluid towards the central cardiopulmonary circulation.23,24 Namely, venous blood can be divided into two ‘compartments’: unstressed volume, which constitutes 70% of volume and represents a sort of a blood reservoir (blood that fills veins to a transmural pressure of 0 mmHg); and stressed volume, which constitutes the remaining 30% of venous blood volume and represents the extra blood volume that elevates wall tension, thereby determining venous return and preload.22,23
During HF decompensation, SNS activation causes a shift from the unstressed compartment (splanchnic veins) to central circulation, thus causing a rapid increase in central venous pressures and consequent development of congestion. In addition to the adverse effects on cardiac and vascular remodelling, which significantly contribute to HF progression, sustained SNS hyperactivity promotes congestion by precipitating inflammation, renin–angiotensin–aldosterone system stimulation, and detrimental effects on renal function, as further discussed.25 In reference to the aforementioned point, it is crucial to emphasise that, akin to the acute setting, renal dysfunction further amplifies SNS activity through afferent signals and renin–angiotensin–aldosterone system, thereby establishing a detrimental feedback loop.26
Increased salt and water retention by the kidneys is a signature of advanced HF. Traditional theories usually ascribed the aforementioned increase to either ‘forward’ or ‘backward’ failure, indicating reduced cardiac output with low kidney perfusion and increased venous pressures favouring transudation, respectively.27,28 However, these concepts were shown to be incomplete, as they largely neglected the contribution of neurohumoral and inflammatory changes, which were shown to be critical for HF progression.29
The inability of the kidneys to excrete salt and water in the setting of HF includes virtually all parts of nephrons.30 First, the deterioration of the glomerular filtration rate is mediated by a reduced number of functional glomeruli, increased angiotensin II levels, increased renal venous pressures and increased intra-abdominal pressures. Second, sodium and water reabsorption in the proximal tubule is facilitated by intrinsic renal mechanisms (increased filtration fraction), neurohumoral changes (increased expression of sodium transporters), increased lymph flow (reduced colloid osmotic pressure in the interstitium) and venous congestion (increased renal interstitial pressures). Third, in the loop of Henle, increased sodium reabsorption is favoured by neurohumoral activation, a reduced amount of filtrate (as a result of increased proximal reabsorption), vasa recta hypoperfusion and renal venous congestion. Fourth, a reduced amount of chloride delivered to macula densa triggers a strong paracrine and neurohumoral response (SNS, renin–angiotensin– aldosterone system, antidiuretic hormone), which jointly contributes to further sodium and water reabsorption. Finally, reabsorption of salt and water is further increased in the distal tubules and collecting ducts, owing to upregulated aldosterone and antidiuretic hormone. It is also worth mentioning that intrinsic renal derangements occurring in HF, which are beyond the scope of this review, already impair natriuresis, even before clinical signs of HF become evident.
The formation of pulmonary and tissue oedema principally occurs consequent to imbalance between hydrostatic and oncotic pressure within the interstitium.31 There are several compensatory mechanisms that prevent oedema formation, despite increased hydrostatic and reduced oncotic pressures. First, glycosaminoglycans (GAGs) gather around a central protein forming proteoglycans.32 The GAGs that are a part of different proteoglycans connect together through hydrogen bonds to form strong, ‘oedema-resistant’ interstitial network.33 In fact, most water molecules in the interstitium are bound to this network, and as long as GAGs are closely associated, a relatively small increase in interstitial pressure will result in a substantial rise in intravascular hydrostatic pressure.34
In contrast, the lymphatic system carries a notable safety factor for oedema prevention both in systemic and pulmonary vascular beds.35 This is elicited by the potency to increase the fluid drainage up to 50-fold in the face of increasing filling pressures, and by massive drainage of interstitial proteins, which causes reduction of colloid osmotic pressure within the interstitium. The above-noted capability of the lymphatic system to prevent oedema formation increases with the duration of increased filling pressures, owing to increased diameter and flow inside the system. Hence, in the acute setting, even a relatively small increase in pressure can overcome colloid osmotic pressure and lead to oedema, whereas in the long term, significantly higher pressure may not manifest evident oedema.36
As HF progresses, sodium saturates the GAG-rich interstitial network, impairing its integrity.37 Namely, as GAGs are polyanionic, they are able to bind large amounts of cations, such as sodium, which at first serves as a protective mechanism against hypernatraemia in HF.34 Yet, long-term saturation of GAGs with sodium changes the function of proteoglycans and destroys the integrity of the interstitial network.37 Consequently, the tissue becomes less resistant to oedema formation. An additional factor that may further promote oedema formation is increased capillary permeability. Such changes in vascular integrity might stem from common HF-related comorbidities, such as diabetes, or by various pro-inflammatory states (e.g. infections), which to some extent explain why these diseases commonly underlie acute decompensated HF.38,39
Although the role of the interstitium mostly explains clinical congestion caused by volume overload, its role in volume redistribution and intravascular congestion is also worth mentioning. In healthy individuals, interstitium holds three- to fourfold more water than the intravascular spaces, and, in a sense, the interstitium determines the intravascular compartment volume.7 For instance, the decline in perfusion pressures (e.g. during blood loss) promotes the movement of fluid across the capillary wall from the interstitial space into the intravascular compartment as a form of a compensatory mechanism.7
With HF progression, owing to previously noted alterations, the ratio of fluid amount between extra- and intravascular space increases, which leads to the establishment of a new dynamic equilibrium; that is, the net accumulation of interstitial fluid provides a mechanism through which rising tissue pressure promotes the expansion of plasma volume. In that way, chronic fluid accumulation underpins the expansion of intravascular volume, thereby providing the ‘foundation’ for the onset of haemodynamic congestion and subsequent symptomatic clinical congestion, which often manifests in recurring cycles of decompensation.
To What Extent Do Biomarkers and Other Clinical Indices Correspond with Congestion Pathophysiology?
Conforming to the multifaceted nature of pathophysiological processes involved in HF decompensation, clinical presentations of congestion can be somewhat stratified based on the predominant mechanism. Specifically, symptoms, such as pitting oedema, rales (indicating pulmonary oedema), ascites and malabsorption (associated with gastrointestinal oedema), primarily arise from volume overload. Conversely, orthopnoea, bendopnoea, prominent jugular veins and the presence of a third heart sound predominantly signify intravascular congestion. An overview of the distinctions between intravascular and extravascular congestion, along with their respective clinical, biochemical and imaging indices, is presented in Figure 1.
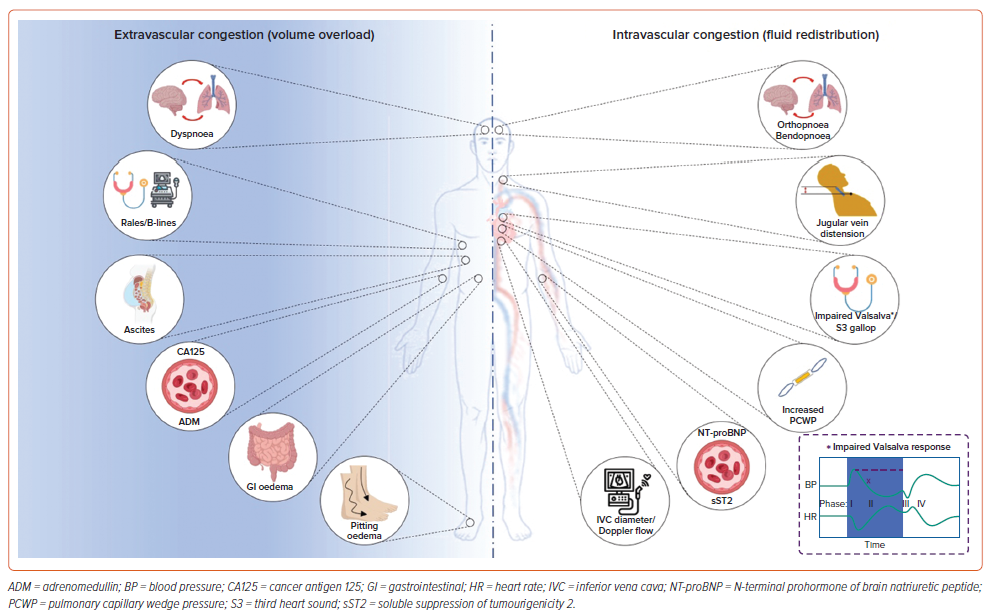
Tissue Congestion
Although the detection of excessive fluid in tissues in the form of pitting oedemas is easily appreciated, the presence of pitting oedema has limited sensitivity and specificity in the establishment of HF diagnosis.40 The presence of basal pulmonary crackles/rales, a classic clinical manifestation of cardiogenic pulmonary oedema, is a valuable indicator of acute HF.41 Conversely, in advanced chronic HF, rales become less evident due to increased compensatory lymphatic drainage.42 Furthermore, assessing residual congestion; that is, after decongestive therapy, by means of physical examination, has proven challenging.40 For instance, alleviation of dyspnoea is an inadequate indicator of decongestion, as patients who remain free of dyspnoea often still exhibit significant clinical or haemodynamic congestion.43,44 In fact, many authors advocate that HF rehospitalisations and consequent poor outcomes often result from inadequate assessment of residual congestion.10 Therefore, alternative approaches for assessing tissue congestion/interstitial oedema, including biomarkers, imaging methods and long-term monitoring devices, have sparked considerable interest in recent years.
CA125, a transmembrane glycoprotein that is widely recognised as a marker of ovarian cancer, has recently emerged as an indicator of volume overload in patients with decompensated HF.11 Specifically, multiple studies showed that CA125 levels are associated with indices of congestion, such as peripheral oedema and serosal effusion, in these patients.45–47
The pathophysiological background explaining increased CA125 levels in HF is still debatable, but available evidence indicates that there are two principal contributors that mutually interact.11 Excessive fluid accumulation in serous tissues elicits mechanical stress, which causes shedding of the extracellular domain of CA125, thus increasing its plasma values.48 Second, low-grade inflammation, a frequent companion of HF, stimulates CA125 production via the Jun N-terminal kinase pathway.49 The principal factors that impede the use of CA125 in congestion management are: long half-life that limits inferring about short-term changes, lack of specificity (elevated in ovarian tumours, pleural effusions, cirrhosis etc.) and as female patients can exhibit higher CA125 levels than men, sex differences need to be taken into account when interpreting it in HF context.
Adrenomedullin (ADM) is a peptide primarily involved in maintaining vascular integrity and permeability barrier function, which might serve as a marker of congestion in acute HF.50 It is hypothesised that in diseases that involve endothelial dysfunction (including HF), ADM might represent a sort of a compensatory mechanism, although currently there is no convincing evidence that ADM is either detrimental or beneficial for patients with endothelial dysfunction.51 Since ADM levels are positively associated with clinical signs and biomarkers of both intravascular (natriuretic peptides, PCWP, orthopnoea, jugular venous pressure; JVP) and extravascular congestion (CA125, interstitial oedema), ADM can be envisioned as a tool for integrated assessment of both types of congestion. However, available data regarding its value in HF monitoring are limited, and ADM lacks standardised assays for measurement, thus generating variability in results across different laboratories.
Apart from the classic chest X-ray, which constitutes a part of the standard workup for suspected acute HF due to its ability to detect pulmonary tissue congestion (although 20% of patients with congestion exhibit a normal chest X-ray), other imaging modalities have also been used to assess congestion.52 For instance, thoracic CT is considered the gold standard for detecting interstitial oedema in the lungs, while lung ultrasound (LUS) is increasingly used, as evidence indicates that LUS findings correlate well with multiple congestion indices.
LUS can be used to quantify the presence of interstitial pulmonary oedema by detecting comet-tail reverberation artefacts called B-lines.53 B-lines are assessed in multiple areas of the lungs, and their sum appears to correlate with both PCWP and radiographic congestion score.53 In addition, it was demonstrated that LUS can predict HF-associated hospitalisation with greater accuracy compared with commonly used clinical, biochemical and imaging parameters.54 Of note, remote dielectric sensing is another non-invasive tool aimed at pulmonary congestion assessment.55 In simple terms, the technology is based on the detection of differences in bioimpedance between air and fluid.55 Preliminary studies indicate that such analysis correlates well with established congestion indices (B-lines, PCWP, natriuretic peptides) and may even predict length of in-hospital stay.56
At first glance, changes in bodyweight seem like an attractive option to assess residual congestion. However, since HF is associated with cardiac cachexia, it is commonly impossible to ‘decipher’ the cause of weight loss.57 Furthermore, cachexia can further exacerbate oedema, as it leads to hypoalbuminemia and consequent reduction in oncotic pressure.58 Accordingly, a decrease in bodyweight does not necessarily lead to improvement of in-hospital or post-discharge morbidity or mortality, albeit an increase in bodyweight seems to portend to worse outcomes.59 Nonetheless, as cachexia is a relatively late complication of HF, bodyweight assessment should not be a neglected component of management of these patients.
Intravascular Congestion
Despite the fact that the intravascular pressure–volume relationship can differ among individuals and clinical conditions, cardiac catheterisation with direct measurement of right atrial pressure and PCWP remains the gold standard for diagnosing intravascular congestion.60 However, the invasive nature of pulmonary artery catheterisation limits its routine clinical use, and despite its ability to improve haemodynamics, the ESCAPE trial showed that its use in guiding decongestive therapy did not improve crude clinical outcomes compared with serial clinical assessment.61
Dyspnoea, a hallmark symptom of HF, can occur regardless of the predominant congestion phenotype, and in fact, its pathophysiology seems to be multifactorial.40 In contrast, orthopnoea, characterised by exacerbated dyspnoea upon assuming a recumbent position, stems from a sudden surge in preload that overwhelms an inadequately adaptive heart.41 Thus, orthopnoea serves as a more indicative marker of intravascular congestion rather than pulmonary oedema. Similarly, bendopnoea, denoting worsening dyspnoea upon bending forwards, is caused by significant positional increase in right- and left-sided filling pressures.62 It is postulated that the mechanism of increased cardiac filling pressure is an exaggerated increase in intra-abdominal pressure that occurs with bending in healthy individuals.62 As bendopnoea is caused by filling pressure increase, most authors envision it as a sign of intravascular rather than tissue congestion.
JVP has been identified as the most valuable clinical indicator for evaluating ventricular filling pressures, and thus for the intravascular congestion assessment.63 Similarly, inferior vena cava diameter and collapsibility is the most common method of echocardiographic right atrial pressure estimation.64 However, the usefulness of JVP in assessing congestion is constrained by notable interobserver variability, and evaluating JVP in obese patients (common in HF) can often be challenging.63
The third heart sound, a low-frequency diastolic sound, is caused by an abrupt limitation of the LV inflow during early diastole that causes vibration of the heart and its contents.64 Although commonly present in HF patients, the third heart sound is often found in young, otherwise healthy, individuals and patients with significant volume overload.64 Since the third heart sound is elicited by a cardiac filling abnormality, it is obvious that it will correlate more with intravascular congestion rather than interstitial oedema.
Rarely used, but a potentially useful clinical sign, is a response to the Valsalva manoeuvre. In a healthy individual, the strain phase of Valsalva is characterised by reduced blood pressure owing to a reduction in preload.65 In patients with HF, blood pressure rises with strain and remains elevated throughout the whole strain phase, as the LV remains adequately filled, despite the drop in preload as a result of elevated filling pressures.65 In fact, such a response was shown to be associated with invasively measured PCWP.66
Despite the limited sensitivity and specificity of each, the presented clinical signs and symptoms appear to be a viable tool for discriminating between the prevailing causes of congestion. Nonetheless, the clinical skills needed to perform physical examination seem to deteriorate in general, presumably owing to routine reliance on advanced diagnostic methodology.40 However, the problem appears to be more significant in the case of low perfusion assessment, for which we have limited tools.40
The principal biomarker for intravascular congestion assessment, and HF in general, are natriuretic peptides.22 Mechanistically speaking, natriuretic peptides are increasingly produced in HF as a consequence of increased ventricular wall stress.67 Although the body of literature concerning the role of N-terminal prohormone of brain natriuretic peptide in HF is extensive, and well beyond the scope of this review, there are several pitfalls worth addressing.
First, serum concentrations of natriuretic peptides exhibit notable intraindividual biological variability, and can be influenced by various cardiac and non-cardiac factors, including genetic predisposition, obesity, age, AF and renal function, all of which may hinder HF-related interpretation.68
Second, while patients experiencing a more substantial reduction in natriuretic peptides after treatment generally have a lower risk of adverse events, the evidence regarding their role in guiding decongestion treatment remains inconclusive.13,69
Third, natriuretic peptides are less affected by right-sided HF and its repercussions.70 Natriuretic peptides hold particularly limited value in HF with preserved ejection fraction, as even normal serum levels are frequently observed in this condition.68
Soluble suppression of tumourigenicity 2 (sST2), a decoy interleukin-33 receptor, emerged as another biomarker of intravascular congestion.71 The mechanism behind the observed increase in serum levels during HF decompensation appears to be associated with the release of proinflammatory cytokines by activated vascular endothelial cells and lung tissue.72 In a sense, sST2 might serve as a form of a bridge between inflammatory and neurohormonal systems.73 Its serum levels are positively associated with both invasively and non-invasively assessed intracardiac pressures.74
Importantly, unlike natriuretic peptides, sST2 levels were shown to correlate well with indices of right-sided HF.75 Recently, sST2 has emerged as a surrogate marker indicating diuretic resistance in patients presenting with acute HF with concomitant renal dysfunction.76 Similar to most putative congestion biomarkers, sST2 interpretation in the context of HF is also burdened by non-specificity, but also certain methodological issues.73 In addition, conclusions regarding the pathophysiological mechanisms that lead to sST2 elevation in HF are not based on hard evidence.
Apart from the previously mentioned inferior vena cava diameter and inspiratory variation assessment, multiple echocardiographic parameters are used to estimate filling pressures with variable accuracy. Left-sided pressures are most commonly estimated based on Doppler tracing of transmitral flow and tissue Doppler of the mitral annulus. Additionally, the diagnostic accuracy of these parameters is significantly enhanced by integrative assessment of LV/left atrial size and function, along with estimated pulmonary artery pressure. Of note, left atrial reservoir strain has recently emerged as an alternative option for assessing left atrial pressure (LAP), particularly in cases where other modalities are not feasible.77 Finally, the evaluation of portal vein flow, hepatic vein flow and renal vein flow might significantly enhance the characterisation of the congestive phenotype in HF understanding.78
The aforementioned techniques are predicated on the premise that venous flow patterns are contingent upon the right heart’s ability to manage venous return. As the right ventricle function deteriorates, forward flow towards the right atrium during systole fades, culminating in systolic reversal evident in pulsed-wave Doppler of the hepatic vein and renal veins. In contrast, in the face of right ventricle dysfunction, portal vein flow changes from monophasic to pulsatile. However, integrative assessment is imperative, as pulsed-wave-based methods have several limitations, including the inability to differentiate between right heart volume and pressure overload. For instance, tricuspid regurgitation may mimic systemic venous congestion (systolic reversal).
The development of small implantable devices has not bypassed the assessment of intracardiac pressures. Currently, there are multiple continuous intracardiac pressure monitoring systems on the market. CardioMEMS, a wireless implantable device for continuous pulmonary artery pressure measurement, has been most extensively studied and was in fact associated with a reduction in hospitalisations due to HF.19 The potential of such sensors resides in the capability to detect changes in pressures before overt HF decompensation, facilitating early treatment adjustments and potentially preventing it. As pressures in the pulmonary artery do not always reflect LAP, the V-LAP system has recently emerged as a method for remote LAP assessment.79
Integrative Assessment: Utilising the Pathophysiological Background to Improve Management of Heart Failure Patients
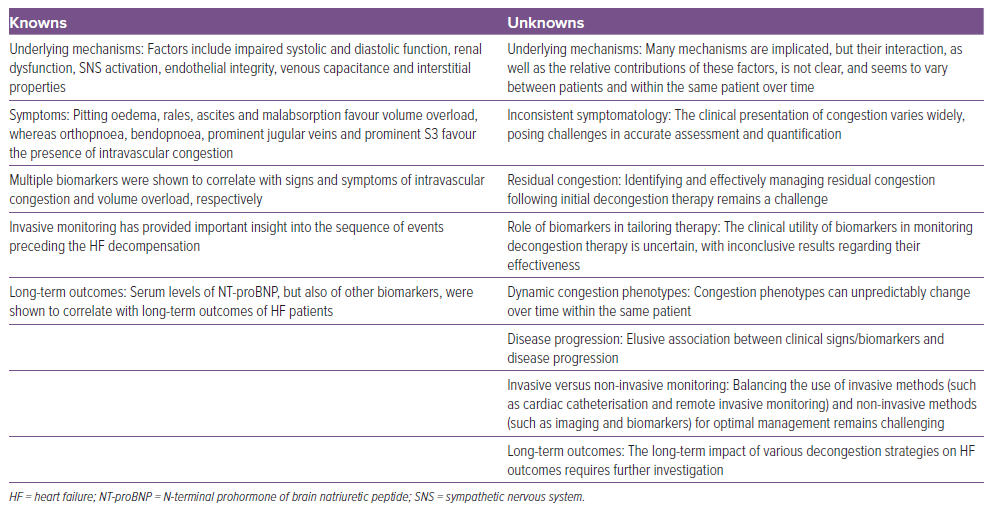
Although in the previous part of this review we dichotomised intra- and extravascular congestion as separate concepts, it is obvious that patients will often present with overlapping features. Moreover, congestion in HF is a changing landscape, and patients may transition to another phenotype any time during the disease course. Therefore, the role of integrated assessment is not to choose between the two but rather to decipher which phenotype prevails and, consequently, in which way could the approach be tailored to improve outcomes. In Table 1, we aimed to summarise the current knowledge and gaps in knowledge pertaining to congestion in HF.
Several authors have proposed algorithms wherein understanding the prevailing phenotype would translate to management decisions. Although congestion phenotypes are optimally assessed invasively, outside the critical care setting, these methods are not feasible. Similarly, although remote invasive monitoring carries low risk and has formidable diagnostic accuracy, its use is currently constrained by high costs and limited availability. Thus, a multiparametric approach, using clinical signs and symptoms, biomarkers, and non-invasive imaging, is proposed to establish the congestion phenotype in everyday clinical practice.
Overall, most authors do not contest that loop diuretics are the first-line treatment, as achieving appropriate natriuresis will alleviate both congestion phenotypes. In contrast, the primary unmet need regarding decongestion therapy is residual congestion, and treatment strategies for it are quite diverse and less supported by conclusive evidence. The principal difference in management between the two phenotypes is envisioned in the fact that patients with volume redistribution as a predominant mechanism will require less aggressive diuretic therapy than those with volume overload.
Boorsma et al. proposed prioritising the resolution of intravascular congestion by achieving proper natriuresis, as intravascular congestion may hinder fluid translocation from tissues to the intravascular space.39 Patients should therefore be re-evaluated for signs of intravascular congestion (presence of bendopnoea/orthopnoea/elevated JVP, inferior vena cava collapse, natriuretic peptides) soon after diuretic initiation. If the symptoms persist, diagnostic evaluation should be steered towards indices of tissue congestion (rales/pitting oedema, CA125, LUS/chest X-ray), whereas treatment should be shifted towards fluid translocation potentially by adding an aquaretic drug.
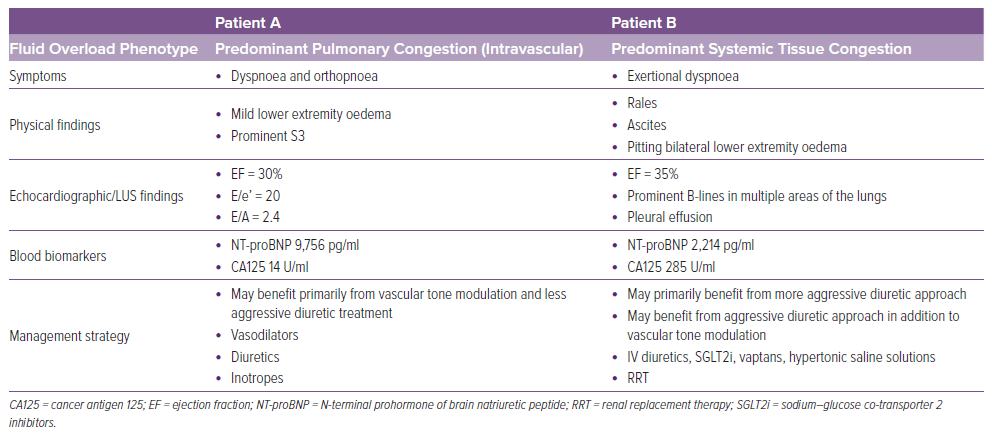
De la Espriella et al. proposed timing at which each evaluation should be performed.10 Except for clinical signs and symptoms, they pointed out that natriuretic peptides and ultrasound should be performed at all four points (at admission, prior to discharge, during transition from hospital to home and during outpatient follow-up), although they addressed that during outpatient follow-up, ultrasound should be used only in selected cases. In contrast, considering the relatively long half-life, they rendered that CA125, an index of extravascular congestion, should not be measured prior to hospital discharge. Finally, the Heart Failure Association has proposed the use of a multiparameter-based evaluation of congestion prior to discharge in a position statement.2 In Table 2, we compared clinical findings between two diametrically opposite putative patients that would benefit from congestion phenotype assessment.
Conclusion
In summary, congestion in HF is commonly mistaken for volume overload, although evidence suggests decompensation can occur without significant water accumulation. The pathophysiology of congestion in HF is complex, involving impaired cardiac function, neurohumoral changes and venous capacitance alterations. The distinction between intravascular and extravascular congestion in the setting of HF often blurs, as patients frequently exhibit overlapping features of both. Moreover, HF congestion is dynamic, with patients transitioning between phenotypes over time. Thus, the focus of integrated assessment is not to prioritise one over the other but rather to identify the prevailing phenotype to tailor management for improved outcomes accordingly.
Various algorithms have been proposed to guide management based on congestion phenotypes, although invasive assessments are optimal, but not always feasible outside critical care settings. Therefore, a multiparametric approach using clinical signs, biomarkers and noninvasive imaging is advocated in routine practice to establish the congestion phenotype.
Distinguishing between volume redistribution and volume overload guides the aggressiveness of diuretic therapy, with a focus on resolving intravascular congestion first to facilitate fluid translocation from tissues. Routine re-evaluation for signs of congestion informs treatment adjustments, with additional modalities, such as natriuretic peptides and ultrasound, employed at different stages of patient care. The timing and selection of assessments are outlined in various proposals, emphasising a comprehensive approach to congestion management.