










The global incidence of heart failure (HF) is increasing, notably HF with preserved ejection fraction (HFpEF), which accounts for over 50% of new diagnoses of HF in the community.1,2 Contemporary data suggest that 900,000 people are currently living with HF in the UK.3 HFpEF is associated with increasing age, and represents the most common HF phenotype in those aged ≥65 years.4 However, the rising incidence of HFpEF cannot be explained by ageing alone, and may also reflect the increasing prevalence of obesity and type 2 diabetes (T2D), two comorbidities strongly associated with HFpEF.5 Furthermore, increasing T2D and obesity in children, adolescents and young adults aged <30 years places these individuals at a higher lifetime risk of developing HFpEF.6 According to Diabetes UK, nearly 5 million people in the UK currently have T2D and a further 14 million are considered at risk.7 Over the past decade, the burden of prediabetes has also risen exponentially, mirroring the epidemiology of T2D. Moreover, 63% of UK adults were classed as overweight or obese in 2020, a number projected to rapidly increase.7 HFpEF is also associated with other comorbidities, such as chronic obstructive pulmonary disease, AF, chronic kidney disease and hypertension.2
HFpEF is increasingly recognised as a heterogeneous syndrome comprising diverse pathophysiological features.8 Although left ventricular (LV) diastolic dysfunction is the predominant feature, additional elements include left atrial (LA) dysfunction, right ventricular dysfunction, focal and diffuse myocardial fibrosis and ventricular–arterial stiffening.9–12
Currently, the pathophysiology of HFpEF remains to be fully elucidated, and the heterogeneity of disease mechanisms and their relative importance at an individual patient level may further confound treatment approaches. Aside from the recent positive evidence base for the use of sodium–glucose cotransporter 2 inhibitors, there remains a lack of treatment options with established prognostic benefit.13 Hence, clinical outcomes in HFpEF remain poor, with HF hospitalisation rates of 24% at 1 year and, following HF hospitalisation, a 1-year mortality rate of 36%.14 Symptomatic HFpEF is also associated with significant socioeconomic costs and a poor quality of life.1
HFpEF is now increasingly recognised as a systemic syndrome, rather than simply a disease of impaired LV diastology and secondary adaptations. In a paradigm shift away from the traditionally ascribed model of excessive afterload inducing diastolic dysfunction, an inflammatory hypothesis of HFpEF has been proposed, placing coronary microvascular dysfunction (MVD) at the core of its pathophysiology.15 In the inflammatory hypothesis, chronic comorbidities are thought to induce a systemic proinflammatory state, resulting in endothelial dysfunction (cardiac and extracardiac) associated with impaired vasodilatory capacity and MVD, which, in turn, is thought to promote downstream sequelae, including hypertrophy, fibrosis and stiffening of the myocardium, as well as LA dilation and abnormal LV relaxation.15
This review explores the importance of MVD in HFpEF pathophysiology. Diagnostic methods for identifying MVD are also examined, followed by a discussion of the reported prevalence and prognostic implications of MVD in HFpEF.
Pathophysiology of Microvascular Dysfunction
Myocardial coronary blood flow (CBF) is regulated by a combination of aortic pressure, epicardial coronary arterial vessel patency and microvascular resistance, factors that can be studied through wave intensity analysis via simultaneous cardiac catheterisation and coronary guidewire Doppler assessments. The phasic compression and decompression of the coronary microvasculature by surrounding cardiac myocytes during myocardial contraction and relaxation further influences coronary flow, so-called ‘cardiac–coronary coupling’.16
In the healthy heart, the majority of coronary flow occurs during diastole driven by ventricular relaxation, with the backward expansion wave (BEW) reflecting decompression of the microvasculature during early diastole. In MVD, during exercise and pharmacologically induced microvascular dilation, blunting of the BEW (which ordinarily accelerates flow) and accentuation of the backward compression wave (which ordinarily decelerates flow) has been noted.17 Therefore, in HFpEF, impaired lusitropy (i.e. the degree of LV relaxation and diastolic dysfunction) may attenuate the BEW and thereby cause impaired myocardial perfusion.18 Furthermore, in phenotypically similar pressure overload conditions such as hypertrophic cardiomyopathy and aortic stenosis, similar disturbances in wave intensity analysis may occur, contributing to MVD.19,20
MVD is defined as an abnormality in the myocardial microcirculation leading to an inadequate vasodilatory response following physiological or pharmacological stress.21 Similar to HFpEF, MVD is in itself a heterogeneous entity, with complex underlying pathophysiological mechanisms that remain incompletely understood. Clinically, in patients with angina, two clear primary endotypes have been defined, namely ‘structural’ or ‘functional’ MVD, based on angiographic studies and blood flow responses to pharmacological vasodilation.18 Although both endotypes exhibit impaired myocardial perfusion reserve (MPR) in response to adenosine, the mechanisms are different. Structural MVD is associated with elevated minimal microvascular resistance; in functional MVD the microvascular resistance remains normal, with reduced vascular tone at rest, but during stress there is attenuated vasodilatory reserve.17,22 Conversely, structural MVD exhibits normal resting myocardial blood flow but with an impaired ability to augment blood flow in response to stress, and diminished peripheral endothelium-dependent dilatation, which can precipitate exercise-induced hypertension. Both endotypes have a similar core phenotype with a high prevalence of inducible ischaemia and inefficient cardiac–coronary coupling during physical exercise. However, their pathogenesis differs at the microvascular and endothelial levels.
Pathophysiology of Microvascular Dysfunction in HFpEF
Microvascular Dysfunction in Antecedent HFpEF
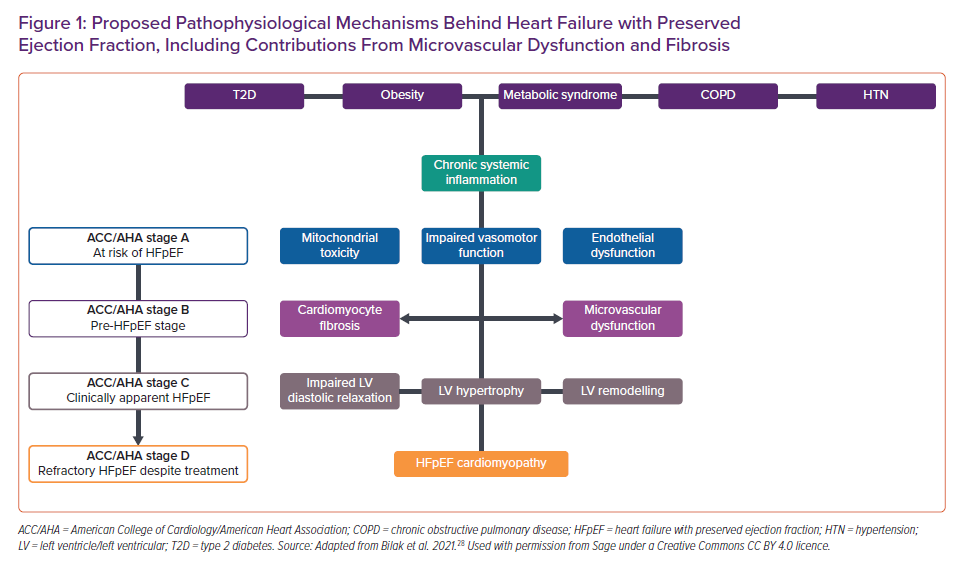
MVD may contribute to the progression of HFpEF and has been proposed as a precursor of HF in otherwise asymptomatic individuals (e.g. those with T2D).23–25 Recognising the clinical and prognostic significance of these early cardiomyopathic changes, the American College of Cardiology/American Heart Association (ACC/AHA) has classified such individuals as having Stage B HF.26 Figure 1 summarises the currently accepted key stages of HFpEF development, including potential contributions from MVD in relation to the relevant ACC/AHA stages of HF.
In a large cross-sectional study of at-risk HFpEF subjects (n=336) comprising women with angina and T2D, Doppler echocardiography was used to investigate MVD.27 Defining MVD as coronary flow reserve (CFR) <2.5, the reported prevalence of MVD was 59%.27 A further 76 patients underwent additional phenotyping with a range of markers of endothelial dysfunction, inflammation and diastolic dysfunction. Seventeen inflammatory biomarkers were negatively correlated with CFR and 15 inflammatory markers were positively correlated with E/e′. Both CFR and E/e′ were only correlated in the subgroup of patients with MVD and signs of increased filling pressure (E/e′ >10; p=0.012), indicating a potential association between inflammation and MVD in T2D-associated HFpEF.27
The clinical syndrome of HFpEF is characterised by severe exercise intolerance.8 In a large cross-sectional study of 247 asymptomatic subjects (mean ± SD age 51.8 ± 11.9 years, 55% men) with T2D and ACC/AHA Stage B HF, the mean reported MPR was reduced compared with that of age- and sex-matched controls (2.60 ± 1.24 versus 3.54 ± 1.15, respectively; p<0.001).24 Reduced MPR (analogous to MVD) was present in the absence of coronary artery disease (CAD) and it corresponded with impaired exercise tolerance test, as expressed by the gold standard peak oxygen consumption.24 Early impairments in peak oxygen consumption have been shown to be one of the first signs of Stage B HF.28 Furthermore, these findings suggest that MVD may be present in otherwise asymptomatic individuals who are at risk of HFpEF in the future.28 Abnormalities of resting and stress blood flow suggestive of MVD have been reported in individuals with metabolic syndrome and obesity (albeit without HF).29,30 Similarly, lower coronary microvascular density (rarefaction) has been reported in adults with severe obesity.31
In a retrospective study of 201 subjects without known HF but comprising a high burden of HFpEF precursors such as T2D, obesity and hypertension, and in whom obstructive CAD was excluded by PET, incident HFpEF was noted in 36 patients during a median follow-up of 4.1 years.32 PET-derived MVD (defined as MPR <2) was noted in 54% of the population. MVD was an independent predictor of HFpEF development and was independently associated with worse LV diastolic function (E/e′).32 These findings of impaired myocardial blood flow in comorbid conditions strongly associated with HFpEF but before the onset of HFpEF are consistent with MVD underlying a range of different HFpEF phenotypes.
Microvascular Dysfunction in Established HFpEF
MVD in HFpEF arises from a combination of microvascular structural alterations (e.g. rarefaction), endothelial dysfunction (endothelium-dependent MVD) and/or vascular smooth muscle dysfunction (endothelium-independent MVD).18 The endothelium is a monolayer of cells that lines the inner vascular wall and plays a key role in the maintenance of vascular homeostasis, initiating and perpetuating inflammatory and immune-mediated responses through the secretion of inflammatory cytokines and proteins.33 Nitric oxide (NO) is crucial to endothelial and smooth muscle function and is synthesised by NO synthase (NOS).22 Patients with functional MVD have a heightened resting microvascular blood flow and NOS reactivity, reflecting a near-maximal vasodilatory state at rest (reduced resting microvascular tone), leading to attenuated vasodilatory responses to stress.17,22
Endothelial NOS (eNOS) dysfunction is thought to be integral in the pathogenesis of HFpEF.15 A systemic inflammatory state induced by comorbidities such as T2D, obesity and chronic obstructive pulmonary disease may lead to the increased production of endothelial reactive oxygen species (ROS).33 The reaction between ROS and eNOS cofactors diminishes NO production and bioavailability. The resulting decline in NO availability reduces levels of protein G kinase, which is essential for the phosphorylation of titin, a cytoskeletal protein responsible for myocardial diastolic recoil and distensibility.34 Therefore, impaired titin phosphorylation due to the dysregulation of the NO–protein G kinase axis may contribute to impaired lusitropy and LV diastolic reserve in HFpEF.34
Biopsy studies in HFpEF have previously demonstrated deficiencies in NO signalling and altered collagen homeostasis, with increased titin phosphorylation.34 This has further been implicated as a potential mechanism underlying the transition from hypertensive heart disease to HFpEF.35 In a murine model of HFpEF, pharmacological inhibition of eNOS enhanced inducible NOS (iNOS) activity, resulting in subsequent dysregulation of protein control within cardiomyocytes, with the accumulation of misfolded proteins, cardiomyocyte dysfunction and, again, impaired lusitropy.36 Importantly, pharmacological inhibition of iNOS also resulted in improved lusitropy and exercise tolerance.36 Although impaired lusitropy has an adverse impact on coronary flow, resulting in ischaemia exacerbated by physiological stress (e.g. exercise), ischaemia may itself precipitate exertion-related diastolic dysfunction.18 Therefore, conclusive ascertainment of causality between MVD, ischaemia and diastolic dysfunction remains challenging.
In humans, autopsy data have previously shown that HFpEF subjects have greater microvascular rarefaction, lower microvascular density and myocardial fibrosis compared with controls of a similar age and regardless of epicardial CAD severity, with the authors proposing microvascular endothelial inflammation as the possible trigger for MVD and fibrotic development.37
Diagnostic Assessment and Prevalence of Microvascular Dysfunction in HFpEF
A range of invasive and non-invasive tests provides information on MVD. At present there is no consensus on the most appropriate diagnostic approach to assess MVD in HFpEF. Existing diagnostic modalities have relative advantages and disadvantages, with a multimodality approach considered optimal. Supplementary Material Table 1 summarises the key studies of MVD in HFpEF across the various modalities.32,38–48
Invasive Assessment
Invasive coronary angiography enables the detection of obstructive epicardial CAD. In addition, the observation of delayed coronary arterial contrast flow ‘slow-flow phenomenon’ may reflect increased coronary microvascular resistance, and therefore underlying MVD. Both the Thrombolysis in Myocardial Infarction (TIMI) frame count (based on the number of cine frames required to opacify the distal coronary arteries; i.e. a reflection of coronary arterial perfusion) and TIMI myocardial perfusion grades are relatively simple, crude and surrogate measures of MVD.47
A more detailed invasive assessment of coronary microvascular function allows dichotomisation of MVD into pathophysiologically distinct structural and functional endotypes.49,50 Structural MVD (i.e. impaired endothelial-independent microvascular vasodilation) is diagnosed by a reduced CFR and/or a high index of microvascular resistance (IMR), whereas functional MVD refers to impaired endothelial-dependent function and is an evaluation of both epicardial or microvascular vasomotor abnormalities. However, both endotypes may also coexist.50
Endothelium-independent function is dependent upon underlying myocyte tone. Both CFR and IMR are measured using thermodilution and/or intravascular Doppler techniques following administration of intravenous vasodilators, such as adenosine. In healthy individuals, CBF ordinarily increases three- to fourfold in response to increased myocardial oxygen requirements. CFR is the ability of the CBF to match the metabolic demand and is measured as the ratio of maximum flow after vasodilator-induced hyperaemia to resting absolute myocardial blood flow. CFR reflects the combined vasodilator capacity of both the epicardial coronary arteries and the microvascular coronary arterioles. The IMR represents the product of distal coronary pressure and hyperaemic mean transit time. Although the IMR is independent of haemodynamic status and coronary flow, concomitant obstructive epicardial CAD may result in its overestimation.51
Endothelium-dependent dysfunction is assessed using intracoronary acetylcholine infusion. In the normal endothelium, acetylcholine induces vasodilatation at both epicardial and microcirculatory levels by stimulating NO synthesis. In MVD secondary to endothelial dysfunction, acetylcholine triggers insufficient NO-mediated vasodilation and/or even paradoxical vasoconstriction.44,49
Dryer et al. evaluated 30 patients with HFpEF (defined as LV ejection fraction (LVEF) ≥50% and clinical HF) in comparison to 14 controls who also underwent cardiac catheterisation.39 Patients with ≥50% coronary artery stenosis were excluded. CFR and IMR were measured after adenosine administration, with thresholds of CFR ≤2.0 and IMR ≥23 to determine MVD. Subjects with HFpEF had lower CFR (mean ± SD 2.55 ± 1.60 versus 3.84 ± 1.89; p=0.024) and a higher IMR (26.7 ± 10.3 versus 19.7 ± 9.7; p=0.037) than the controls.39 Within the HFpEF group, over one-third (36.7%) had overt MVD (i.e. both abnormal CFR and IMR), and a similar number had either reduced CFR or raised IMR. The authors concluded that these findings indicate the presence of different vascular subtypes of MVD, and hence that MVD may be a heterogeneous entity.39
In a larger study of 106 HFpEF patients who underwent invasive coronary angiography (and cardiac MRI [CMR] in nearly half), obstructive CAD was present in over 50% of participants.44 MVD was highly prevalent: 85% overall and 81% in those without obstructive CAD. Endothelial-independent MVD (defined as CFR <2.0 and/or IMR ≥25) was noted in 66% of participants, whereas endothelial-dependent MVD (defined as an abnormal coronary vasoreactivity response to acetylcholine) was observed in 24%.44 Of those who also underwent CMR (n=52), 27% had evidence of previous MI and 30% had extracellular volume (ECV) >30%, indicative of diffuse fibrosis.44
Yang et al. combined echocardiography with invasive angiographic assessment of coronary physiology with Doppler flow wire and intracoronary adenosine to assess MVD in 162 consecutive HFpEF patients.48 Endothelium-independent MVD was defined as CFR ≤2.5 and endothelium-dependent MVD was defined as an increase in CBF ≤10% in response to acetylcholine. Overall, MVD was present in 72% of patients; isolated endothelium-dependent MVD was present in 29% of patients, with isolated endothelium-independent MVD in 33% and combined MVD in 10%. HFpEF patients with endothelium-independent MVD had lower diastolic relaxation velocities (mean ± SD 7.0 ± 1.8 versus 8.4 ± 2.9 cm/s; p=0.002) and higher estimated filling pressures (E/e′ 13.1 ± 4.1 versus 9.6 ± 3.4; p<0.001) than those with normal endothelial function, suggesting a pathophysiological link between impaired lusitropy and impaired myocardial perfusion.48
In summary, invasive studies demonstrate that the prevalence of MVD in HFpEF is consistently high, ranging between 70% and 85% depending on the diagnostic thresholds used, which vary between studies: CFR ≤2 to ≤2.5, IMR ≥23 to ≥25.39,44,48 The high prevalence of epicardial CAD is not surprising given the burden of comorbidities associated with both atherosclerosis and HFpEF. As epicardial CAD affects myocardial perfusion, it is difficult to evaluate the relationship between HFpEF and MVD in cohorts with highly prevalent epicardial CAD. CFR is also typically calculated in a single coronary vessel (commonly the left anterior descending artery [LAD]), assuming this is representative of the myocardium globally.52
Published studies also differ in their definitions of HFpEF (ranging from normal LVEF and impaired diastolic relaxation to fulminant HF as defined by Framingham criteria).39,48 Moreover, in currently published data, sample sizes are relatively small, with poor matching of clinical cohorts. This is primarily due to the opportunistic nature of the studies, in which both study subjects and controls were derived from a pool of patients referred for invasive angiography for a clinical indication (including symptoms of CAD and HF).
Non-invasive Assessments
Echocardiography
Transthoracic echocardiography (TTE) is readily available and remains the imaging cornerstone for HFpEF diagnosis. TTE can detect abnormalities of LV function (impaired LV diastolic relaxation and LV strain) and geometry (LV hypertrophy and remodelling), as well as increased LA volume, abnormalities of right ventricular function and raised pulmonary pressures, all of which form part of the diagnostic criteria for HFpEF.53 In addition, myocardial perfusion can be studied through Doppler evaluation of coronary flow, typically by interrogation of the LAD, which can be visualised from conventional parasternal short axis views. Measurement of diastolic coronary flow velocities (CFV) thus enables calculation of the CFR (i.e. the ratio of CFV at rest and after hyperaemia).
Doppler echocardiography for assessment of CFR is validated against invasive angiographic Doppler guidewire measurement (r=0.94, mean difference 0.10 ± 0.18) and PET-based CFR (limits of agreement [–0.75, 0.71]; within-subject coefficient of variation 11% and reliability 0.84).54,55 However, the limitations of echocardiography include CFR assessment being confined to a single epicardial coronary artery (typically the LAD) and a reliance on adequate imaging windows, which may be suboptimal in the setting of HFpEF, especially in the presence of concomitant lung disease and obesity.
In a small study of 77 HFpEF patients in whom obstructive CAD was excluded, MVD (defined as CFR <2) was present in 66%.43 Furthermore, patients with impaired CFR had an increased E/e′ ratio (13.5 ± 4.1 versus 9.7 ± 3.6, p<0.001), LV mass index and mass to volume ratio when compared with HFpEF patients without MVD. In multivariate analysis, MVD was found to be an independent predictor of impaired exercise capacity as assessed by the 6-minute walk test.
The largest prospective study to date assessing MVD in established HFpEF, namely PROMIS-HFpEF, recruited 202 patients with a mean HFA-PEFF score of 6.1.45 In that multicentre study, the mean age of subjects was 74 years, with a slight female predominance (55%), and the vast majority had multiple comorbidities, clinical characteristics highly typical of HFpEF. Importantly, subjects with significant CAD on the basis of positive stress testing, invasive angiography or unrevascularised CAD were excluded. CFR was measured with adenosine-stress transthoracic Doppler echocardiography in the LAD territory. Using this methodology, 75% of patients were identified as having MVD (defined as CFR <2.5).45
PET
PET is regarded as the gold standard for perfusion quantification. PET-derived data offer further insights into the phenotypes of MVD through assessment of myocardial blood flow or perfusion reserve.46 MPR is a surrogate measure of the vasodilatory capacity of small vessels and an accepted surrogate marker of MVD, if epicardial CAD is excluded.56
Srivaratharajah et al. studied a large patient cohort (n=376) with normal LVEF (≥50%) and no prior CAD, in whom global and regional LV myocardial flow reserves were calculated using 82Rb PET.46 Seventy-eight subjects had HFpEF (defined by LVEF ≥50% and clinical signs of HF), and comparisons were made with hypertensive (n=112) and non-hypertensive subjects (n=186). Mean ± SD MPR was significantly lower in HFpEF subjects (2.16 ± 0.69) compared with both hypertensive (2.54 ± 0.80; p<0.02) and normotensive (2.89 ± 0.70; p<0.001) subjects. MVD (MPR <2) was present in 40% of HFpEF subjects. Moreover, a diagnosis of HFpEF was associated with 2.62-fold greater unadjusted odds of having global impairment in MPR and remained a significant predictor of reduced global MPR after adjusting for the presence of comorbidities.46 However, the retrospective nature of the analysis of patients referred for clinical assessment of CAD based on symptoms of angina or exertional breathlessness is a major limitation of that study.
Cardiac MRI
CMR offers multiparametric imaging options that, in addition to the evaluation of MVD, allow for a comprehensive assessment of myocardial structure, function and enhanced tissue characterisation.57 In the clinical evaluation of HFpEF, CMR has emerged as a key tool for the aetiological workup of suspected HFpEF.58
In a single-centre prospective study of suspected HFpEF patients (n=154) undergoing standard evaluation with TTE, the addition of stress perfusion CMR identified new, clinically significant pathologies in over one-quarter of patients (27%), including hitherto undiagnosed CAD (ischaemia or infarction), hypertrophic cardiomyopathy, constrictive pericarditis and MVD (assessed qualitatively based on global perfusion detects).59 These findings indicate that the use of CMR may facilitate more targeted, disease-specific therapies in HFpEF. CMR also further enables stratification of HFpEF into pathophysiological subtypes (e.g. right ventricular dysfunction, LA dysfunction, focal and diffuse myocardial fibrosis) known to influence prognosis.9–11,60
CMR also permits quantitative perfusion assessment with greater spatial resolution than that of PET. In a study of 19 patients with symptomatic HFpEF (LVEF threshold >45%) and in whom LV hypertrophy was absent and CAD excluded by CT coronary angiography, MVD (defined as MPR <2.5) was present in the most (13/19).42 Mean ± SD MPR was lower in HFpEF patients than in controls (2.29 ± 0.62 versus 3.38 ± 0.76, respectively; p<0.05). Interestingly, subjects with HFpEF also had increased myocardial ECV, a quantifiable surrogate marker of diffuse fibrosis, compared with controls (0.29 ± 0.04 versus 0.25 ± 0.04; p<0.05). Although myocardial fibrosis has been related to MVD, the exact relationship between the two entities has not yet been fully elucidated, and further investigation is required.
Another CMR study used phase contrast imaging of the coronary sinus to evaluate global myocardial blood flow in 25 patients with HFpEF, 13 patients with hypertensive LV hypertrophy (no HF) and 18 control subjects.61 HFpEF was defined by LVEF ≥50%, evidence of diastolic impairment on echocardiography (E/e′ ≥15) and elevated natriuretic peptides (B-type natriuretic peptide >200 pg/dl). Mean CFR differed significantly across the HFpEF, hypertensive LVH and control groups (2.21 ± 0.55 versus 3.05 ± 0.74 versus 3.83 ± 0.73, respectively; p<0.001). MVD, based on CFR <2.5, was observed in 19 of 25 HFpEF patients.61 In addition, CFR was independently correlated with B-type natriuretic peptide concentrations (β=−68.0; 95% CI [−116.2, −19.7]; p=0.007), suggesting a role for MVD as an imaging marker of disease severity.
In a recently published prospective longitudinal study, 101 HFpEF patients underwent CMR perfusion imaging at our centre (Leicester NIHR Biomedical Research Centre, Glenfiedl Hospital), as well as intense phenotyping with rigorous clinical evaluation, multiple plasma biomarker profiling and TTE.38 The mean ± SD age of the patients was 73 ± 9 years and the mean ± SD LVEF was 56 ± 5%. Importantly, subjects with known epicardial CAD or CMR evidence of CAD (i.e. infarction or regional perfusion defects) were excluded from further perfusion analysis. Subjects underwent multiparametric CMR, including adenosine stress and rest perfusion with absolute quantitation of myocardial blood flow during stress and at rest, and evaluation of both focal late gadolinium enhancement (LGE) imaging and diffuse myocardial fibrosis (ECV; T1 mapping technique). MVD, defined as MPR <2.0, was present in 70% of patients. MPR was lower in HFpEF subjects than in controls (1.74 ± 0.76 versus 2.22 ± 0.76, respectively; p=0.001) and independently predicted adverse outcomes. Resting blood flow did not differ significantly between the HFpEF subjects and controls.38 In contrast, two much smaller CMR studies (n=19 and n=25) reported elevated resting blood flow in patients with HFpEF.42,61 CMR-derived MPR was also correlated with echocardiographic (E/e′; r=–0.34, p=0.002) and biochemical (B-type natriuretic peptide; r=–0.22, p=0.038) markers of disease severity.38
Relationship of Fibrosis to Microvascular Dysfunction in HFpEF
Although the evidence for the presence of MVD in HFpEF is consistent, the relationship between MVD and fibrosis, which is also frequently reported, is less well understood. Myocardial fibrosis is a common endpoint of many cellular processes in HFpEF.37,62 Our own work has previously shown that diffuse fibrosis, as determined by ECV, is highly prevalent in HFpEF, is correlated with indices of LA and LV remodelling and is predictive of clinical outcomes.11 Similarly, prior autopsy data from HFpEF patients (n=124) revealed a greater burden of myocardial fibrosis (median area 9% versus 7%) and lower microvascular density (a surrogate for MVD attributed to microvascular rarefaction; median 961 versus 1316 vessels/mm2) compared with age-matched controls (n=104).37 Furthermore, myocardial fibrosis was shown to be inversely correlated with MVD in both controls (r=−0.28, p=0.004) and HFpEF patients (r=−0.26, p=0.004), suggesting a pathophysiological link between myocardial fibrosis and MVD.37
However, our findings from CMR differ from those of the aforementioned autopsy study and a much smaller CMR study.38,42 Our results highlight no significant linear correlation between MPR and diffuse fibrosis (r=−0.10, p=0.473), and no difference in MPR in those with and without focal fibrosis (mean difference −0.03; 95% CI [−0.37, 0.3]).38 This may be explained by the less advanced stage of HFpEF seen in our cohort compared with the end-stage disease profile encountered in the autopsy patients who also had a higher prevalence of CAD (65%).37 Furthermore, there are inherent differences in the assessment of MVD and fibrosis between invasive and non-invasive modalities. In the small CMR study of HFpEF (n=16), a significant negative correlation between MPR and ECV was reported (r=−0.54, p=0.002).42 Further studies are required with serial imaging to gain insights into the relationship between the myocardial fibrosis and perfusion, including rarefaction.
In summary, the evidence from both invasive and non-invasive imaging studies suggests that MVD is highly prevalent in HFpEF. MVD is likely to be a heterogeneous entity, and its pathophysiological contribution to the development of HFpEF may vary across different HFpEF phenotypes. The complex relationship between fibrosis, vascular rarefaction and MVD in the pathophysiology of HFpEF remains incompletely understood, and prognostic studies with well-phenotyped patients may shed more light.
Prognostic Implications of Microvascular Dysfunction in HFpEF
To date, outcome data pertaining to the role of MVD in HFpEF prognosis is limited to only a handful of studies and is devoid of PET-based data. In a single-centre retrospective study of 162 patients, in which obstructive CAD (>50% stenosis of any coronary artery or prior acute coronary syndrome) was excluded, the presence of invasive coronary Doppler-flow-wire-detected MVD showed a signal towards worse outcomes during long-term follow-up (median 12.5 years).48 Compared with preserved endothelial function, endothelium-dependent MVD revealed a trend towards worse mortality (adjusted HR 2.81; 95% CI [0.94–8.34]), albeit statistical significance was not reached (p=0.06). However, endothelium-independent MVD did show a significant association with mortality (adjusted HR 3.56; 95% CI [1.14–11.12]; p=0.03).48
The above findings are contrast with those of another invasive prospective multicentre study with a smaller sample size (n=106), comprising both obstructive and non-obstructive CAD, substantially shorter follow-up (median 18 months) and limited by fewer events (n=45).44 Although patients with obstructive CAD were noted to have more adverse events during follow-up than those without obstructive CAD (74% versus 46%, respectively), the presence of invasively detected MVD overall, nor stratification according MVD phenotype (endothelial-dependent or endothelial-independent), did not exhibit any significant association with prognosis.44
In a prespecified exploratory analysis of the multicentre PROMIS-HFpEF study (n=201, 1-year follow-up), the presence of echocardiographically detected MVD (CFR <2.5), compared with no MVD, was associated with significantly higher incidence rates of adverse events across a range of endpoints, including the composite of cardiovascular death and/or recurrent HF hospitalisations, the composite of all-cause death and/or first HF hospitalisation and recurrent but not first all-cause hospitalisation.64 However, that study was not powered for outcomes and the observed event rates were also very low. For example, the outcomes of cardiovascular death or first HF hospitalisation only occurred in 15 participants (four deaths).63
To date, the strongest body of evidence linking MVD in HFpEF with prognosis comes from CMR data. In a single-centre observational study of 163 HFpEF patients who underwent CMR for the clinical indication of screening for myocardial ischaemia and in whom a prior history of MI was excluded, the presence of MVD was strongly associated with adverse outcomes (cardiovascular death or HF hospitalisation) during a median follow-up of 4.1 years.40 MVD, as defined by CFR <2.0 and derived from phase contrast cine imaging of the coronary sinus, was prevalent in 42% of HFpEF patients who experienced adverse events, compared with 3% of patients without adverse events.44 Furthermore, MVD outperformed both focal fibrosis detected by LGE (0.881 versus 0.768; p=0.037) and global longitudinal strain (0.881 versus 0.747; p=0.036) in predicting events.40 However, these findings are limited by the retrospective study design, involving patients referred for clinical CMR and ischaemia evaluation, which may have introduced bias.
Further supportive evidence for CMR-derived MVD as a prognostic marker in HFpEF comes from our own study.38 In that prospective cohort study of 101 HFpEF patients, strengthened by the presence of an age- and sex-matched control group (n=43), patients were followed for a median of 3.1 years. Using a different method (validated against PET) using model-independent deconvolution of myocardial signal intensity curves to quantify absolute myocardial blood flow, MVD was defined as MPR <2.0.64 MVD was independently predictive of the composite endpoint of all-cause mortality and/or HF hospitalisation, after adjustment for clinical, biochemical or imaging parameters.38 In a subgroup analysis of the previously mentioned multicentre study of HFpEF (overall n=106), comprising only a limited number of adverse events, 52 patients underwent CMR.44 The group with a reduced MPR index (a surrogate for MVD) was noted to have more adverse events compared with the group with normal MPR, reinforcing the strong signal for the role of MVD in prognosis.
Although, MVD shows promise as a prognostic marker in patients with HFpEF, there is a paucity of studies evaluating outcomes in patients recruited with rigorous HFpEF criteria. To date, studies have assessed MVD outcomes in heterogeneous cohorts of patients, often with comorbidities that are independently associated with adverse prognosis.66 In particular, T2D, which is highly prevalent in HFpEF and typified by the presence of microvascular disease, has been shown to be associated with worse prognosis in HFpEF.65 Chronic comorbidities such as obesity, T2D and hypertension commonly coexist in HFpEF and are likely to contribute to MVD through varying mechanisms and compound the risk of adverse events in HFpEF.66–68
Furthermore, sex and ethnic differences in outcomes and prognosis in HFpEF have been described.69,70 Therefore, further studies are needed to evaluate the true prognostic implications of MVD in HFpEF, with due consideration given to specific HFpEF phenotypes, sex and ethnicity.
Conclusion
The global incidence of HFpEF is increasing. There is a close association between chronic diseases and the pathophysiology and prevalence of HFpEF. Although the prevalence of MVD in HFpEF appears to be approximately 75% and it is independently associated with adverse prognosis, there is a relative lack of direct evidence for its pathophysiological role in HFpEF. Nonetheless, the studies available to date paint a consistent picture strongly linking MVD to the development and progression of HFpEF. Further studies are needed to assess whether measures of MVD (invasive and non-invasive) can be used to assess disease progression in HFpEF or, indeed, whether MVD is a potential treatment target. Clinical trials targeting fibrosis and perfusion in the HFpEF population are an attractive avenue for further research.
Click here to view Supplementary Material.
Clinical Perspective
- Heart failure with preserved ejection fraction (HFpEF) is an increasingly common form of heart failure with only one proven drug class of effective therapy.
- Microvascular dysfunction is an important pathophysiological mechanism in the development of HFpEF and is associated with adverse clinical outcomes.
- Both invasive techniques and cardiovascular imaging play a key role in the evaluation and management of HFpEF, including assessment of myocardial perfusion and fibrosis.