







Heart failure is currently one of the leading causes of death and disability worldwide, which makes it a major public health problem.1,2 Traditionally, heart failure is considered a complex syndrome with several features, including abnormal myocardial function and excessive, continuous neurohumoral activation. In this context, the current optimal pharmacological treatment of heart failure focuses on the suppression of neurohumoral activation as well as on the regulation of the fluid volume overload, haemodynamics and optimisation of heart rate control.3 However, there is accumulating evidence indicating that additional mechanisms, such as inflammatory activation and metabolic impairment, are also involved in the pathogenesis of heart failure; this evidence has stimulated the search for novel therapeutic strategies.
More than half a century ago, Richard Bing, who is often called the ‘Father of Cardiac Metabolism’, emphasised that the heart is more than a pump, it is also an organ that needs energy from metabolism.4,5 At that time, the necessity of metabolic therapy for metabolic diseases was declared. Today, multiple myocardial metabolic abnormalities in heart failure have been revealed; the heart of a patient with heart failure can be described as ‘an engine out of fuel’.6 Moreover, besides myocardial metabolic failure, systemic (peripheral) metabolic regulation has been found to contribute both to major symptoms of heart failure and to disease progression.7
In recent years, a number of promising therapies modulating the cardiac metabolism have been tested. At present, a clinically important question was raised – is there a place for metabolic modulators in the current treatment of heart failure? This article aims to review the rationale and evidence base of metabolic modulators, in particular trimetazidine, in the management of patients with heart failure.
Cardiac Energy Metabolism in Heart Failure
The heart is an organ with a high-energy demand. To perform continuous contractile activity, the myocardium hydrolyses more than 6 kg of adenosine triphosphate (ATP) daily.8 In normal conditions about 95 % of cardiac energy is obtained through the production of ATP from mitochondrial oxidative metabolism, while the remaining 5 % originates from glycolyticATP production.The changes in cardiac energy metabolism in heart failure are complex and depend on the stage and the cause of the heart failure.9 Mitochondrial oxidative metabolism is impaired in heart failure, resulting in a decrease in ATP and phosphocreatine levels in the failing heart. The decrease in glucose oxidation is more critical than changes in fatty acid oxidation.10 The proportion of ATP derived from mitochondrial fatty acid oxidation exceeds that derived from glucose oxidation, but more oxygen is required to produce ATP from fatty acid oxidation compared with glucose oxidation. In response to reduced oxidative metabolism the glucose uptake and glycolysis are elevated. High glycolysis rates and low glucose oxidation rates can result in an increase in the uncoupling of glycolysis from glucose oxidation, leading to the production of lactate and protons, which decreases the efficiency of the heart (see Figure 1).11
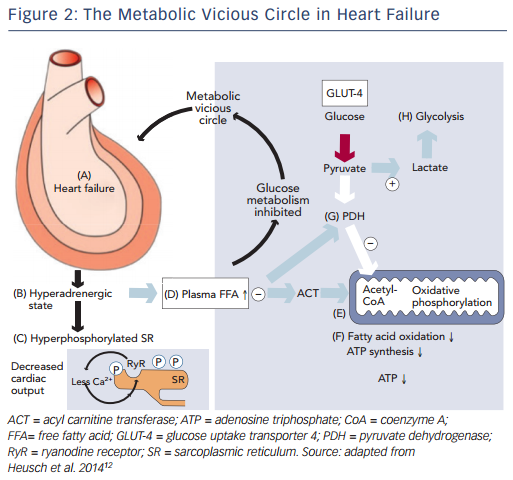
Recently, impaired mitochondrial oxidative metabolism in heart failure was defined using the term ‘metabolic remodelling’, as one component of a broader and more general concept of remodelling covering haemodynamic, neurohumoral, metabolic and inflammatory processes, causing changes in cardiomyocytes, endothelium, vascular smooth muscle cells as well as interstitial cells and matrix.12 This understanding of the processes of remodeling in heart failure has again drawn attention to the so-called metabolic vicious circle, which was proposed by Opie.13 The metabolic vicious circle includes the following sequence of events: the dilation of the myocardium in heart failure (A) leads to the adrenergic activation (B), that in turn hyperphosphorylates the sarcoplasmic reticulum (C) and increases the concentrations of circulating free fatty acids (D); free fatty acids inhibit mitochondrial function at the level of acyl carnitine transferase (E), thus inhibiting fatty acid oxidation and synthesis of ATP (F); plasma free fatty acids also inhibit pyruvate dehydrogenase (G) to promote anaerobic glycolysis (H), rather than oxidative metabolism (see Figure 2). This approach allows for consideration of therapies that target the cardiac metabolism as add-on therapy along with conventional treatment for heart failure.
Cardiac Energy Metabolism as a Potential Target for Therapy in Heart Failure
There is emerging evidence demonstrating that therapeutic regulation of cardiac metabolism by reducing fatty acid oxidation and/or increasing glucose oxidation may be an effective treatment for heart failure.11 Several pharmacological agents that modulate fatty acid metabolism by decreasing the supply of fatty acids to the heart, inhibiting fatty acid uptake and -oxidation or stimulating glucose oxidation have been developed (see Figure 1).
Both beta () blockers and nicotinic acid can decrease circulating free fatty acid levels and, therefore, indirectly reduce myocardial fatty acid oxidation and promote glucose use. blockers are fundamentally important in modifying the course of systolic heart failure.3 It is believed that inhibition of carnitine palmitoyl transferase-1 (CPT-1) activity, increased glucose oxidation and increased efficiency of oxygen use for ATP production may also be partially responsible for the beneficial effect of blockers in heart failure.14 Nicotinic acid, as a broad-spectrum lipid-regulating agent, reduces the frequency of cardiovascular disease events,15 but no additional benefit was found when used in conjunction with an intensive statin therapy.16 Unlike statins, there is no evidence that nicotinic acid reduces the incidence of heart failure in patients with coronary artery disease.
Peroxisome proliferator-activated receptor (PPAR) and PPAR agonists (fibrates and thiazolidinediones) also decrease the circulating free fatty acid supply to the heart, which results in reduced cardiac fatty acid oxidation rates. Fibrates lower the risk of major cardiovascular and coronary events compared with placebo, but do not affect the risk of cardiovascular or all-cause mortality.17 These lipid-regulating agents are not considered to be able to prevent the development of heart failure either. Thiazolidinediones are used to treat patients with type 2 diabetes; however, they may cause worsening of heart failure and increase the risk of heart failure-associated hospitalisation.18 Therefore, thiazolidinediones should not be used in patients with heart failure.3
Etomoxir and perhexiline, which are Inhibitors of CPT-1, decrease the activity of this rate-limiting enzyme for fatty acid -oxidation and thus limit fatty acid oxidation while favouring glucose oxidation (via glucose-fatty acid cycle, called the Randle Cycle).19 Etomoxir was initially developed as an antidiabetes agent and was then shown to improve left ventricular performance in animal studies.20,21 Etoximor was associated with improved exercise capacity but also increased liver transaminase levels in patients with heart failure.22 For this reason, etomoxir is not considered as a suitable modulator for use in heart failure. Perhexiline was initially developed as antianginal drug. Perhexiline inhibits the cardiac, but not the hepatic, isoform of CPT-1 and is associated with improved exercise capacity and left ventricular ejection fraction (LVEF) in patients with heart failure.23 In the late 1980s perhexiline was withdrawn worldwide, with exception of Australia and New Zealand where it remains licensed for the treatment of refractory angina.
Malonyl coenzyme A decarboxylase (MCD) inhibitors increase cardiac malonyl coenzyme A (CoA) levels, which inhibit CPT-1, thereby reducing mitochondrial fatty acid uptake. MCD inhibition leads to increased glucose oxidation, decreased fatty acid oxidation and improved insulin sensitivity.9 Pharmacological MCD inhibitors are under development for the treatment of myocardial ischaemia24 and obesity25. At present, no MCD inhibitors are available for clinical use.
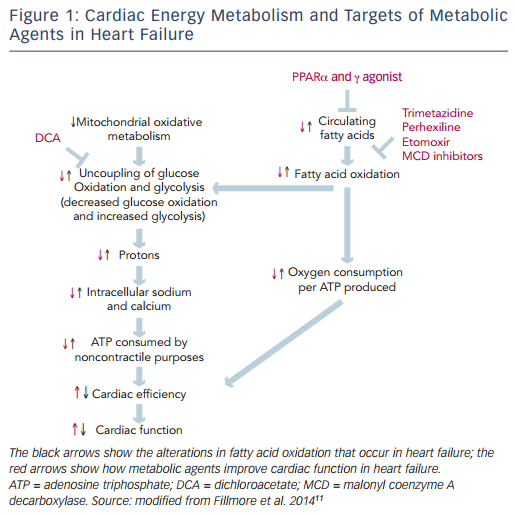
Dichloroacetate (DCA) increases the activity of the mitochondrial pyruvate dehydrogenase complex by inhibiting pyruvate dehydrogenase kinase, and thereby increasing glucose oxidation. In an experimental study DCA was shown to reduce the rate of progression of left ventricular hypertrophy to heart failure.26 Currently, there is no clinical evidence that supports using DCA in heart failure.
Trimetazidine is a partial inhibitor of long-chain 3-ketoacyl CoA thiolase, the key enzyme in the -oxidation pathway. Trimetazidine shifts the myocardial energy metabolism from fatty acid -oxidation towards glucose oxidation, thereby increasing ATP generation and, ultimately, improving contractile function. Trimetazidine has been approved in more than 80 countries worldwide as an antianginal agent. Today, a vast amount of data confirm the efficacy of trimetazidine in heart failure. Ranolazine is similar in structure to trimetazidine and can inhibit fatty acid oxidation; however, this effect is much less pronounced.27 The main mechanism of action of ranolazine in myocytes is the inhibition of the late sodium current. Ranolazine is currently approved as an antianginal agent in USA and Europe. The impact of ranolazine on heart failure has only been investigated in a few clinical trials.28–30 Ranolazine has been shown to significantly increase left ventricular ejection fraction in patients with systolic and diastolic heart failure. The RanolazIne for the Treatment of Diastolic Heart Failure (RALI-DHF) study revealed that ranolazine improves measures of haemodynamics; however, there were no significant effects on relaxation parameters or N-terminal pro–B-type natriuretic peptide concentration in patients with heart failure with preserved ejection fraction.30 The value of this agent in the treatment of heart failure remains to be clarified.
The list of new therapies targeting cardiac metabolism is constantly expanding. Studies of mitochondria-targeted peptides (Szeto–Schiller peptides [especially SS-31] and coenzyme Q10), manganese superoxide dismutase mimetics, hormone replacement therapy, iron chelators and so on attract particular attention. Further experimental and clinical studies are required to confirm their efficacy in heart failure.
Clinical Benefit of Trimetazidine in Patients with Heart Failure
At present, trimetazidine is the only pharmaceutical modulator of cardiac metabolism, and is widely available in clinical practice. In addition to the approved indication (i.e. the symptomatic treatment of stable angina), there has been growing evidence that trimetazidine prevents ischaemia–reperfusion injury after myocardial revascularisation procedures and improves cardiac function in heart failure.
The beneficial effect of trimetazidine in heart failure has been attributed to shifting energy production from fatty acid oxidation to glucose oxidation, which leads to an increased production of high- energy phosphates as well as an improvement in endothelial function, reduction in calcium overload and free radical-induced injury, and inhibition of cell apoptosis and cardiac fibrosis with further beneficial effect on myocardial viability.31–35
The trimetazidine-induced beneficial effect on left ventricular function in patients with heart failure has been shown to be associated with an improvement of the cardiac phosphocreatine:ATP ratio by 33%, indicating the preservation of myocardial high-energy phosphate levels.36 Moreover, the observation that this beneficial effect is also paralleled by a reduction in the whole-body rate of energy expenditure indicates that this effect may be mediated through decreased metabolic demand in peripheral tissues.37 It was found that trimetazidine improves the functional capacity in patients with heart failure when used in conjunction with exercise.38 This positive effect on functional capacity could be explained by the cytoprotective mechanism exerted by trimetazidine on skeletal muscle integrity.39 It is important that trimetazidine acts without affecting heart rate and blood pressure.
Numerous clinical trials have demonstrated the efficacy of trimetazidine in improving New York Heart Association (NYHA) heart failure class, exercise tolerance, quality of life, LVEF, cardiac volumes, and inflammation and endothelial function in patients with ischemic cardiomyopathy.40–46 There are considerably fewer studies that have examined the efficacy of trimetazidine in patients with heart failure of non-ischaemic aetiology.47,48 Nevertheless, trimetazidine has been shown to significantly improve cardiac function and exercise tolerance in patients with idiopathic dilated cardiomyopathy and has extracardiac metabolic effects such as increase in high-density lipoprotein levels and reduction of blood insulin and C-reactive protein levels.
Interestingly, trimetazidine has potential electrophysiological properties. Several studies have demonstrated the beneficial effects of trimetazidine on the parameters of heart rate variability, P-wave duration and dispersion and changes in QT interval that are considered markers of the increased risk of cardiac arrhythmias and sudden cardiac death in patients with heart failure.49–52
In recent years, the ability of trimetazidine to reduce the rate of all- cause mortality in patients with heart failure has become a subject of particular interest. This intriguing observation was made in a number of randomised controlled trials (RCTs) and retrospective cohort studies.
The ability of trimetazidine to improve survival rates in patients with ischaemic cardiomyopathy and multivessel coronary artery disease was first evidenced by El-Kady et al. in a single-centre, open-label, randomised trial.53 In this study, 200 patients were randomised to receive trimetazidine or placebo for 24 months. After 2 years of treatment survival rates were 92 % for the patients treated with trimetazidine versus 62 % for the patients in the placebo group.
In another open-label study, Fragasso et al. randomised 45 patients with heart failure to either conventional therapy plus trimetazidine or conventional therapy alone, with a mean follow-up of 13 months.45 It was noted that, apart from the patients who died during the follow- up period, patients randomised to conventional therapy alone had a higher incidence of cumulative cardiovascular events compared with the patients randomised to trimetazidine.
In the 48-month extension phase and post-hoc analysis of a single- centre, open-label, randomised Villa Pini d’Abruzzo trimetazidine study, 61 patients with heart failure were randomised to receive trimetazidine in addition to conventional treatment or to continue their usual drug therapy for 4 years.54 This analysis showed that, in comparison with conventional therapy alone, the addition of trimetazidine significantly reduced the rate of all-cause mortality by 56 % (p=0.005) and heart failure hospitalisation by 47 % (p=0.002), and improved patients’ functional status (NYHA class and 6-min walking test) and LVEF.
The positive long-term effect of trimetazidine on the reduction of mortality rates was also demonstrated in another single-centre, open-label, randomised trial.55 The results of this study showed a significant effect of trimetazidine modified release in postmyocardial infarction patients with angina and heart failure in terms of a 15 % reduction in the all-cause mortality rate over the 6-year follow-up period (p<0.05) and of major cardiovascular events such as cardiac death, nonfatal myocardial infarction, acute stroke, need for coronary revascularisation, hospitalisation for unstable angina or heart failure (by 15.7 %; p<0.05).
The results of these studies are in line with those reported by Fragasso et al. in a large, international, multi-centre retrospective cohort study involving 669 patients with heart failure (including 362 patients receiving trimetazidine).56 It was noted that the addition of trimetazidine to conventional therapy is associated with a significantly reduced all-cause (by 11.3 %; p=0.015) and cardiovascular mortality rate (by 8.5 %; p=0.050), as well as, a reduction in hospitalisations for any cardiovascular cause (by 10.4 %; p<0.001).
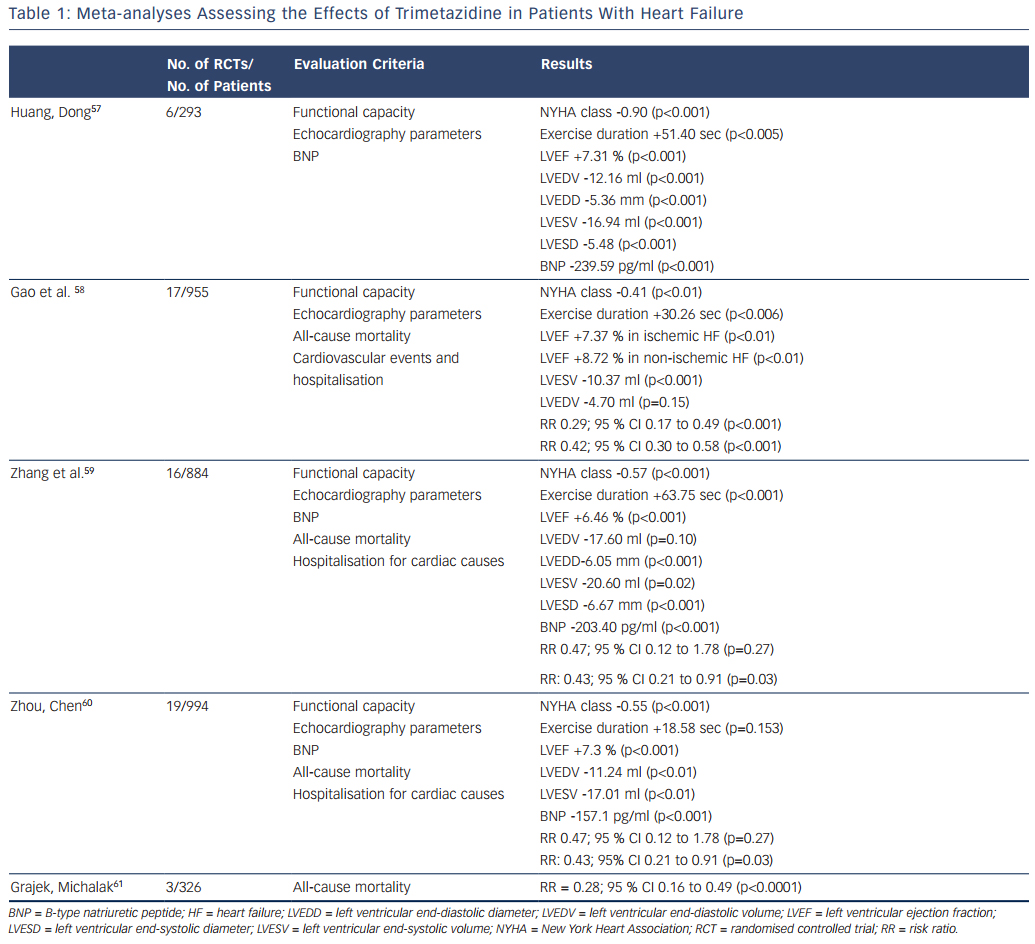
Several meta-analyses of RCTs have been performed to estimate the effects of trimetazidine treatment in patients with heart failure. Table 1 summarises the characteristics and key results of these meta-analyses.
The first meta-analysis was performed by Huang and Dong and included data for 293 patients with heart failure from four RCTs and two crossover design trials.57 Huang and Dong demonstrated that, compared with the control group, trimetazidine reduced the NYHA class and B-type natriuretic peptide (BNP) level, increased exercise duration, improved the indices of cardiac function and quality of life.
Later, Gao et al. published a meta-analysis that pooled data from 17 RCTs, which included 955 patients with heart failure.58 In comparison with placebo, trimetazidine treatment was associated with NYHA class reduction, increased exercise tolerance and improvement of LVEF in patients with heart failure of both ischaemic and non-ischaemic aetiology. The most interesting finding was that in patients with heart failure the use of trimetazidine reduced the rates of cardiovascular events and hospitalisations, and all-cause mortality.
In a meta-analysis performed by Zhang et al., data from 16 RCTs with 884 patients with heart failure also showed the ability of trimetazidine to decrease NYHA class, increase exercise tolerance, improve LVEF and decrease left ventricular end-systolic and end-diastolic diameters, as well as the level of BNP.59 As in the meta-analysis by Gao et al.,58 Zhang et al. noted that trimetazidine reduced the rate of hospitalisation for cardiovascular reasons in patients with heart failure, but not the all-cause mortality rate.
An updated meta-analysis by Zhou and Chen that included data for 994 patients with heart failure from 19 RCTs confirmed the reduction of NYHA class, cardiac volumes and BNP level and improvement in LVEF in patients treated with trimetazidine.60 Again, a reduction in the rate of hospitalisation for cardiac causes was observed. However, there were no significant differences in exercise duration and all- cause mortality rates between patients treated with trimetazidine and those receiving placebo.
Recently Grajek and Michalak presented a meta-analysis evaluating the effect of trimetazidine on all-cause mortality rate in patients with heart failure.61 A total of 326 patients from three RCTs were analysed: 164 who received trimetazidine in addition to a pharmacological heart failure therapy and 162 controls. The results again showed a significant reduction in all-cause mortality rate among patients with heart failure treated with trimetazidine.
The main limitation of these meta-analyses is that they were based on under-powered studies. The number of patients with heart failure included in these meta-analyses was relatively small. Other limitations include the differing designs of the included studies and wide variations in follow-up durations.
The limitations of meta-analyses and retrospective trials should not outweigh other proven facts, such as the ability of trimetazidine to relieve symptoms, improve the quality of life and increase the functional capacity of patients with heart failure. Presently, much importance is given to these parameters; the current European Society of Cardiology guidelines for the diagnosis and treatment of heart failure consider them as important targets in the management of patients.3 Taking into account the above-mentioned finding and also the evidence that trimetazidine delays or reverses left ventricular remodelling in patients with heart failure, we can expect that this agent will be added to the guidelines on the management of patients with heart failure. Moreover, the first steps have already been taken: today, trimetazidine is already included in a number of national guidelines as an agent for treatment of patients with heart failure of ischaemic aetiology.62–64
Trimetazidine can be recommended to patients with heart failure of ischaemic aetiology used in addition to an angiotensin-converting enzyme (ACE) inhibitor (or an angiotensin receptor blocker if ACE inhibitors are not tolerated), a -blocker and a mineralocorticoid receptor antagonist to eliminate the symptoms, increase the functional capacity, normalise haemodynamic parameters and provide a possible reduction in the risk of death and re-hospitalisation.
Conclusions
Increased understanding of the role of systemic and cardiac metabolism impairments in heart failure not only generates new pathophysiological concepts, but also stimulates the search for new therapies for patients with heart failure. Trimetazidine effectively regulates cardiac energy metabolism by reducing fatty acid oxidation and increasing glucose oxidation and its potential should be considered for add-on therapy with conventional treatment of heart failure.