










According to the current universal definition, heart failure (HF) is a clinical syndrome with signs and symptoms secondary to a functional and/or structural cardiac abnormality corroborated by the elevation of natriuretic peptides (NP) or objective evidence of cardiogenic pulmonary or systemic congestion.1 It is typically classified based on the left ventricular ejection fraction (LVEF) in HF, with reduced ejection fraction (HFrEF) when LVEF is ≤40%, HF with mildly reduced ejection fraction (HFmrEF) when LVEF is 41–49%, HF with preserved ejection fraction (HFpEF) when LVEF is ≥50% and HF with improved ejection fraction (HFimpEF) when LVEF was below 40% at baseline but has improved more than 10% and is above 40% with treatment.1
Although the global incidence seems to be stabilising, HF prevalence continues to increase, mainly attributed to population ageing and greater survival with new therapies both for HF and ischaemic heart disease.2,3 Currently, it is estimated that 1–3% of the population is affected, with a projected 46% increase by year 2030.2–4 Over the years, the availability of therapies to treat HFrEF has increased;5–7 for many years β-blockers, angiotensin-converting enzyme inhibitors/angiotensin receptor blockers (ACEI/ARB) and mineralocorticoid receptor antagonists (MRA) remained the cornerstone of treatment. However, since 2014, with the results of PARADIGM-HF that led to the introduction of the angiotensin receptor-neprilysin inhibitor (ARNI) sacubitril/valsartan and the later DAPA-HF and EMPEROR-Reduced leading to the addition of the sodium–glucose cotransporter 2 inhibitors (SGLT2I), a real revolution in the management of HF began.8–10 SGLT2Is are also the first therapy to consistently benefit patients with HFpEF, HFmrEF and HFimpEF.11,12 We now know that the combination of four medications (a β-blocker + MRA + ARNI + SGLT2I) – now referred to as foundational therapy or guideline-directed medical therapy (GDMT) – can dramatically improve the prognosis of patients with HF.
Despite these great achievements in the treatment of HF and the consistent benefits seen in large randomised clinical trials, the implementation of GDMT remains suboptimal.13 In this review, we aim to summarise the evidence behind the implementation of foundational therapy for HF, to tackle some of the fears and misconceptions that contribute to the lack of implementation of newer therapies and to discuss strategies to offer all patients with HF the best treatment available in a timely manner with a focus on HFrEF.
Progress in the Treatment of Heart Failure
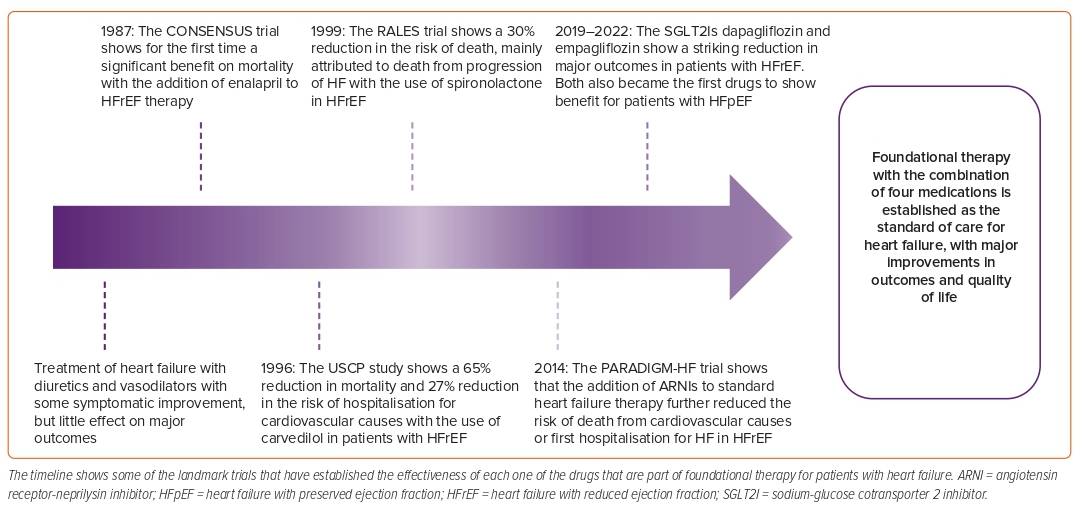
HF was once a deadly disease; in the early years, most patients were treated with diuretics and digitalis and – although there was some symptomatic improvement – the impact of these therapies on prognosis was minimal.14 In 1987, the CONSENSUS trial showed for the first time a striking benefit on mortality of patients with HF with the addition of enalapril to standard therapy; later, other trials confirmed the benefit of renin–angiotensin–aldosterone system inhibition (RAASI) with ACEI/ARB in HF.15–17 In 1996, the USCP Study showed the benefit of carvedilol compared with placebo in patients with HF and an LVEF <35%, with a 65% reduction in the risk of mortality, a 27% reduction in the risk of hospitalisation for cardiovascular causes and a 38% risk reduction in the combined endpoint of hospitalisation or death, which was also later confirmed for both metoprolol and bisoprolol.18–20 In 1999, the RALES study tested the efficacy of spironolactone in patients already treated with ACEI on the basis that RAAS is only transiently inhibited with ACEI/ARB therapies in many patients with HF and that aldosterone levels are still detected in blood, even with adequate dosing of RAASIs, contributing to fibrosis and HF progression.21 RALES showed a 30% reduction in the risk of death in the spironolactone arm, which was mainly attributed to a lower risk of death for progression of HF and sudden death from cardiovascular causes.21 Notably, many of these trials were stopped early because of the greater benefit seen in the intervention arms, demonstrating the consistent benefit of these therapies.
For many years, these therapies constituted the cornerstone for the treatment of HFrEF and, after these landmark trials, there was a period of quiescence without newly available drugs. In 2014, the PARADIGM-HF trial showed that, in patients with HFrEF, treatment with ARNI compared with enalapril caused a 20% reduction in the composite endpoint of death from cardiovascular causes or first hospitalisation for HF.8 In the PARADIGM-HF trial, all patients were required to be on a stable ACEI dose equivalent to at least 10 mg of enalapril daily prior to study entry and to have elevated NP levels. This might explain – in part – why some of the first guideline recommendations on the use of ARNIs reserved the treatment for patients who were still symptomatic, with an LVEF ≤35% on background therapy with β-blockers, ACEI/ARBs and MRAs and with good tolerance to RAASI; later the benefit proved to be consistent for patients naive to ACEI and with lower NP.8,22–24
Some of the most notable medications recently added to the armamentarium in this war against HF are the SGLT2Is. Both dapagliflozin and empagliflozin reduced the risk of the composite endpoint of worsening HF or cardiovascular death in 25% of patients with HFrEF and in approximately 20% of patients with HFpEF, showing a consistent benefit across the spectrum of LVEF (HFrEF, HFmrEF, HFpEF) and becoming the first line of treatment for patients with preserved ejection fraction.9–12,25 Recent meta-analyses have shown that this foundational therapy can reduce the risk of all-cause mortality, cardiovascular mortality and HF hospitalisation (HFh) by nearly 70% in patients with HFrEF, and it is the best treatment available for these patients (Figure 1).26
Suboptimal Implementation of Guideline-directed Medical Therapy for Heart Failure
Although the benefit of GDMT in patients with HF is clear, real-world data show that implementation of these therapies is far from optimal. The CHAMP-HF registry included 3,518 patients with HFrEF and analysed the patterns of GDMT use.13 Overall, <2% of the population had an absolute contraindication to receive ARNI/ACEI/ARB, β-blocker or MRA. Of those who were eligible to receive GDMT, 73.4% received ARNI/ACEI/ARB, 67% a β-blocker, and only 33.4% received an MRA; of those receiving RAASI, only 13% received an ARNI and the rest ACEI/ARB. Less than 30% of the patients receiving ARNI/ACEI/ARB and a β-blocker received target doses, while of those receiving an MRA, >75% were on target dose. Only 1.1% of the patients were on target doses of a combination of three medications.13
More recently, the EVOLUTION-HF registry showed that there is a significant delay in the initiation of novel therapies (dapagliflozin and sacubitril/valsartan) within the 12 months after a hospitalisation for HF. In addition, discontinuation rates were significantly high for ARNI/ACEI/ARBs, β-blockers and MRAs, with a higher persistence rate for dapagliflozin.27 Up-titration to guideline-recommended doses was also lower for ARNI/ACEI/ARBs, β-blockers, and MRAs; since SGLT2Is require no titration, most patients received the target dose.27 Contemporary data from the GWTG-HF registry showed that in the US, only 20% of patients eligible for initiation of SGLT2I received the medication after hospitalisation for HF – even those with diabetes or chronic kidney disease (CKD).28
When evaluating the reasons for the low implementation of therapies for HF, there might be two opposite, extreme misconceptions that may – at least in part – contribute (Figure 2).

The first of these misconceptions is the misconception that HF can reach a ‘stable’ state and that some patients are ‘not so sick’ and, therefore, do not need all therapies. HF is a progressive disease in which persistent neurohormonal activation is one of the main contributing factors for progression.29 Patients with HF, especially those without adequate treatment, have progressive fibrosis, remodelling and dysfunction – even during periods of good functional status and low symptom burden.30 It has been well demonstrated that, in those patients with HF who have an improvement of their LVEF and are asymptomatic, stopping GDMT is associated with worse outcomes, suggesting that what is keeping those patients in a good state is medical therapy.31 In addition, it is likely that initiating low doses of two components of foundational therapy in a symptomatic patient with HF that can, for example, produce vasodilation, offload the left ventricle and reduce sympathetic overstimulation, will lead to an improvement of symptoms that may be misinterpreted by clinicians as a therapeutic success, avoiding the proper up-titration and initiation of other therapies that would also be of great benefit.
Lack of treatment intensification/progression in a patient who is not yet on evidence-recommended doses, or the failure to modify therapy according to guidelines when indicated, is what is known as clinical/therapeutic inertia and is a substantial problem in HF.32,33 Considering that a patient has only ‘mild’ HF because LVEF is not ‘very’ reduced is also a frequent misconception because HF is a clinical syndrome that presents within a spectrum of ejection fraction; for every phenotype there is an increased risk of disease progression and adverse cardiovascular events without adequate treatment.6
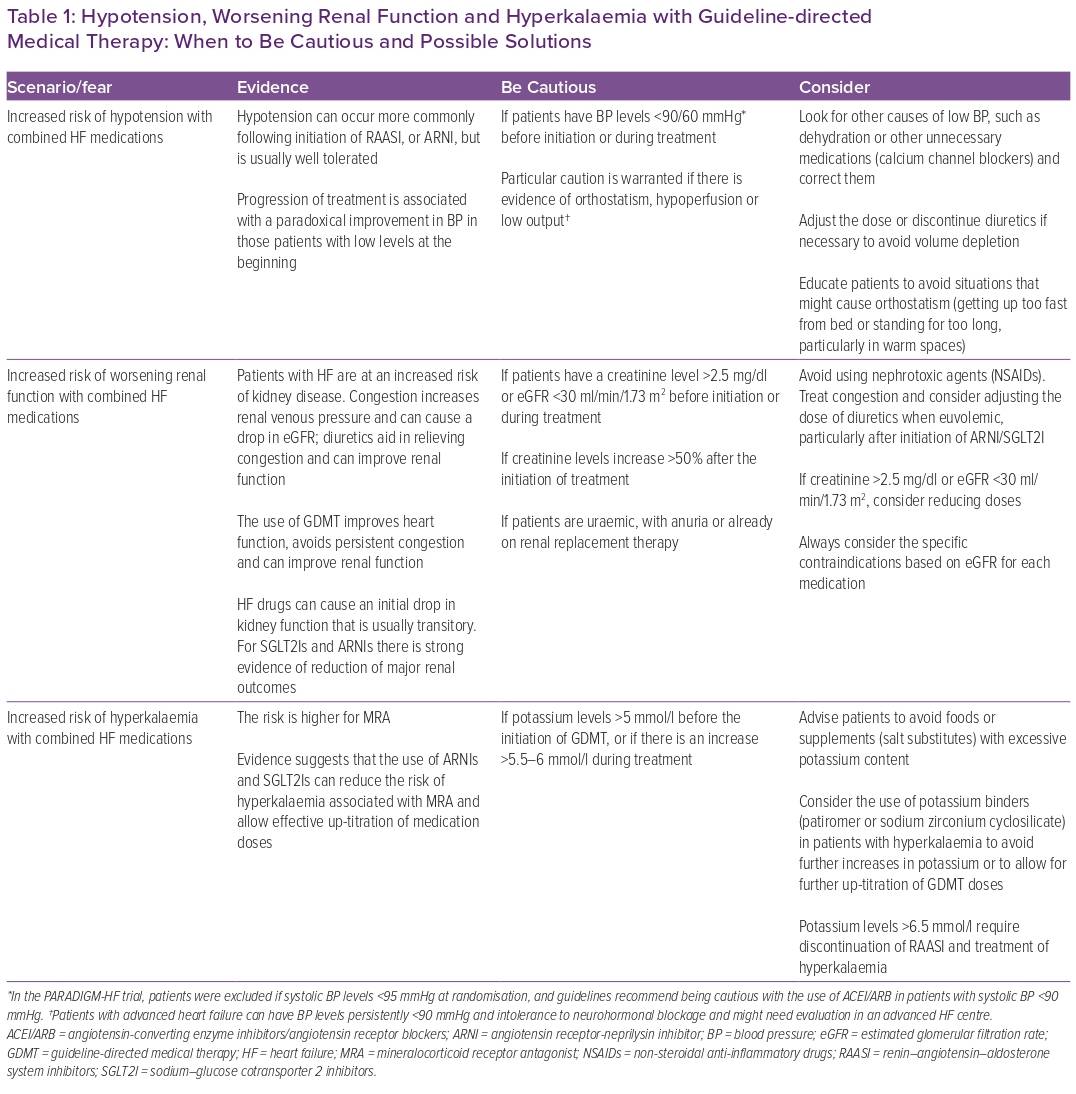
The second of these misconceptions is that some patients may be perceived as ‘too sick’ and probably won’t tolerate the whole treatment. This is especially true for those patients who are or have been recently admitted for an episode of acute decompensated HF, received inotropes during admission, had decreased renal function or hyperkalaemia and those with blood pressure (BP) levels that are in the low to normal limit. Some concerns in these scenarios are that the initiation/up-titration of GDMT might worsen renal function, cause hyperkalaemia or produce hypotension. In fact, some registries suggest that BP and renal function are common factors that determine whether or not a patient is started on medications.13,27
In patients with HFrEF, the left-ventricular pressure–volume loop is displaced to the right, and the end-systolic pressure–volume relationship is shallower than in a normal heart, suggesting that the maintenance of an acceptable stroke volume is dependent on an increased end-diastolic volume and that the left ventricle in HF is quite sensitive to increases in afterload.34 Medications used in HF can produce vasodilation or reduce heart rate, therefore impacting afterload. Contrary to what is thought, reducing afterload in HF helps to reduce the resistance imposed over a failing left ventricle and improves stroke volume.34,35 This is supported by evidence showing a paradoxical BP response in patients with HF and low BP prior to the initiation of some therapies. For MRAs, ARNIs and β-blockers, there is a gradual increase in BP levels for those patients who initiate treatment with systolic BP <105–110 mmHg, while patients who initiate treatment with systolic BP >135–140 mmHg show a reduction after treatment initiation.36–38 In fact, this paradoxical effect on BP seems to be comparable between ARNIs and enalapril, which is frequently the alternative offered to patients who are not started on sacubitril/valsartan.38 The effect of SGLT2Is on BP levels is minimal; however, this paradoxical response is also present.39 So, unless BP levels are <90–100/60 mmHg, there is evidence of orthostatism, hypoperfusion or true low output and all other possible causes have been ruled out and/or managed, concerns regarding BP should not be a reason to withhold, delay or interrupt GDMT.
In patients with congestion, excess intravascular volume involves the venous system and is transmitted to the renal veins.40,41 The kidneys – contained in a non-distending capsule – experience an increase in pressure secondary to venous congestion that reduces renal blood flow and estimated glomerular filtration rate (eGFR). This can lead to activation of tubuloglomerular feedback with the release of renin and consequent activation of the RAAS system, leading to increased sodium and water reabsorption and enhancing sympathetic activation, resulting in more congestion and creating a vicious cycle.42 Decongestion with diuretics decreases intrarenal pressure and improves renal haemodynamics.41,43 Contrary to what is thought, adequate implementation of GDMT in patients with HF will help to improve cardiac performance and avoid re-congestion, thus reducing the risk of worsening renal function.41
Progression of renal dysfunction is a common feature of progressive HF, and the slope of decrease in eGFR is steeper in patients with HF than in healthy people. There are some factors associated with acute alterations in glomerular haemodynamics that can cause a drop in eGFR, such as congestion, low cardiac output, increased intra-abdominal pressure and initiation of RAASIs. The influence of these factors on eGFR is usually transitory and not associated with a loss of functional nephrons. On the other hand, chronic activation of the RAAS system is associated with a loss of functional nephrons and progression of kidney disease, highlighting the relevance of implementing appropriate GDMT for patients with HF.41,44 It is common to see a drop in eGFR in the first weeks after initiation of GDMT;45,46 for most patients, this is transitory, and after some weeks there is usually an improvement in renal function and the slope of progression of kidney disease is slowed.41,45,46 For ARNIs and SGLT2Is, there is strong evidence showing that the use of these therapies is associated with a significant reduction in the risk of progression to end-stage kidney disease, and the benefits seen in relevant outcomes are maintained in patients with lower eGFR.41 Therefore, the fear of worsening renal function should not be a reason to withhold, delay or interrupt GDMT, unless the patient is uraemic, there is a significant increase (usually double) in creatinine level after the initiation of treatment or there is a specific contraindication based on eGFR according to each medications prescribing information.
Hyperkalaemia is common in patients with HF, usually associated with the use of RAASI and related to worse outcomes. When it occurs, a common response is to reduce or stop HF medications, an action that can have harmful effects.47 There is an incorrect perception regarding the combination of MRA and ARNI and the risk of developing hyperkalaemia. A sub-analysis of patients receiving MRA in the PRADIGM-HF trial showed that, compared with patients receiving enalapril, those receiving ARNI had a reduction in potassium levels over time. The risk of hyperkalaemia was similar between groups; however, severe hyperkalaemia (defined as potassium levels >6.0 mEq/l) was more frequent in patients receiving enalapril.48 An analysis of the DAPA-HF trial showed that the incidence of hyperkalaemia, particularly severe hyperkalaemia, was lower in the dapagliflozin arm compared with placebo.49 In the EMPEROR-Reduced trial, patients receiving empagliflozin were 22% less likely to discontinue treatment with MRAs compared with those in the placebo arm.50 Table 1 summarises evidence regarding hypotension, worsening renal function and hyperkalaemia associated with the use of GDMT, with some warnings and possible ways to address these scenarios.
The different components of the foundational therapy for HF act synergistically, somehow protecting each other from causing the development of adverse events, and allowing a faster and safer up-titration of medications. In addition, their benefit is additive, and the combination of these four medications represent the best (better) treatment available.
Initiation and Up-titration of Guideline-directed Medical Therapy: Faster, Stronger!
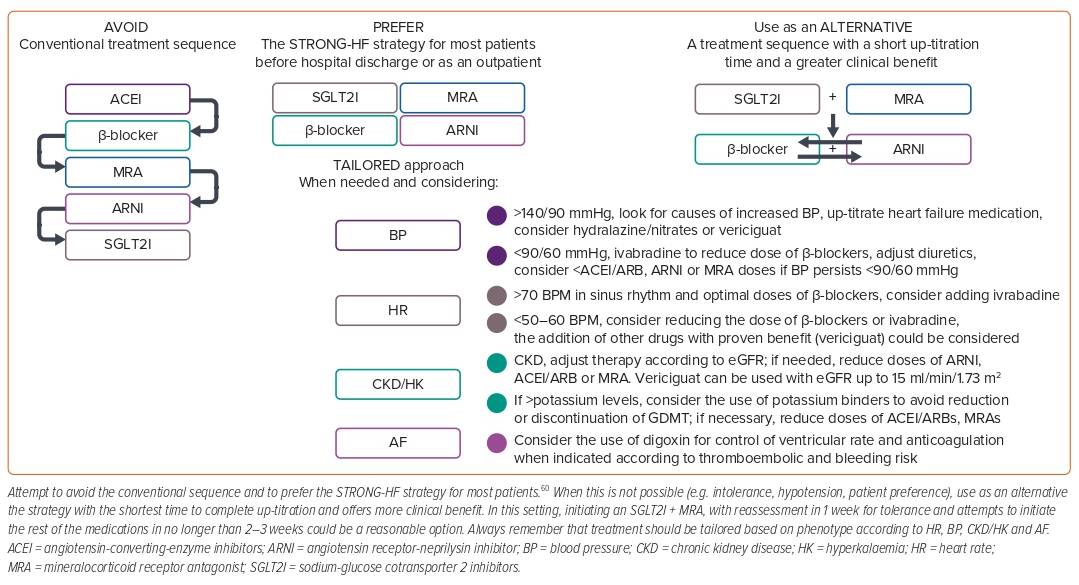
Prior guidelines recommended an escalating algorithm for the treatment of HFrEF.22 In this algorithm, patients were started on low doses of a β-blocker and ACEI/ARB and up-titrated to maximum tolerated doses and then re-evaluated; those who were still symptomatic and with an LVEF ≤35% had an indication to receive an MRA and to up-titrate this to maximum tolerated doses.22 Only those patients who were still symptomatic and had an LVEF persistently ≤35% after maximal doses of β-blockers + ACEI/ARBs + MRAs and who tolerated high doses of ACEI/ARBs were eligible for switching ACEI/ARBs to sacubitril/valsartan.22 Considering that specific recommendations in those guidelines suggested that initiation should be at low doses and up-titration should also be slow, it could take at least 4–6 weeks for a patient to complete one step of the algorithm and then be re-evaluated to move to the next one.22 For each component of GDMT, the benefit seen in major outcomes (mortality, HFh) appears within days to weeks (specifically 19 days for MRAs, 28 days for SGLT2I, 27 days for ARNI and only a few weeks for β-blockers), so delaying the initiation and up-titration of medications comes at the cost of increasing the risk of preventable deaths and HFh.8,52,53 Another issue with the algorithm was that if a patient achieved clinical ‘stability’ and ejection fraction improved >35%, the progression of treatment could be stopped and ‘no further action’ was required.22 This implied stopping treatment with only a few medications at low doses for some patients, contributing to clinical inertia.
In a network meta-analysis, Tromp et al. showed that treating patients with a combination of only an ACEI + β-blocker was associated with a 31% reduction in the risk of all-cause mortality, a 16% reduction in the risk of cardiovascular mortality and HFh and a 36% reduction in the risk of cardiovascular mortality.26 The ACEI + MRA + β-blocker combination was associated with a reduction of 48%, 42% and 53% for each outcome, respectively. Replacing ACEIs with ARNIs increased the benefit in every outcome, but the greatest benefit was seen with the ARNI + MRA + β-blocker + SGLT2I combination, with a risk reduction of 61%, 64% and 67% for each outcome, respectively.26 Thus, stopping treatment prematurely means not providing the whole benefit seen with complete foundational therapy. Other estimates show that, in patients with HFrEF, the lack of initiation, titration or persistence of a β-blocker is associated with an increase of 35% in the relative risk of all-cause mortality and 19–24% in all-cause mortality or hospitalisation; for MRAs, a 24–35% increase in the RR of all-cause mortality and 35–42% in HFh; for ARNIs, a 25% increase in all-cause mortality and 30% in cardiovascular mortality or HFh; and for SGLT2Is, an increase of 13% in all-cause mortality and 31% in HFh.54 For all these reasons, current guidelines eliminated the escalating algorithm and now recommend the initiation of therapy for all eligible patients with HFrEF;6 however, despite these strong recommendations, implementation of newer therapeutic algorithms in the real world remains far from optimal.
Hospitalisation for an episode of decompensated HF is always bad news for patients in terms of prognosis; however, for clinicians, it represents a great opportunity to implement life-saving therapies to reduce the risk of rehospitalisation or death. Initiating HF medications in patients before discharge is associated with a higher likelihood of adherence and allows for closer monitoring during the first doses; in addition, multiple trials have shown that starting HF therapies during hospitalisation is safe and associated with better outcomes.55 The PIONEER-HF trial showed that among patients with HFrEF hospitalised for a decompensation, initiating sacubitril/valsartan before discharge was associated with a greater reduction of N-terminal pro-brain NP (NT-proBNP) compared with enalapril, while rates of worsening renal function, hyperkalaemia, hypotension or angioedema were similar between groups.23 Analysis of secondary exploratory outcomes showed that the initiation of sacubitril/valsartan was associated with a significant reduction in the composite of rehospitalisation for HF and cardiovascular death.56 The EMPULSE trial showed that the initiation of empagliflozin in patients admitted for decompensated HF who were clinically stable was associated with a greater clinical benefit and was safe and well tolerated.57 Smaller studies demonstrated that the use of empagliflozin as a decongestive strategy in high doses (25 mg) early during the decongestion process (12 hours after admission) was associated with a significant increase in urine output, higher decrease in NT-proBNP and greater diuretic efficiency while being safe and well tolerated.58 Initiation of MRAs during hospitalisation for decompensated HF is also associated with a significantly lower risk of mortality, cardiovascular death, HFh and renal failure compared with non-initiation or discontinuation.59 Based on these data, the 2021 guidelines for the treatment of HF recommended that all patients should be initiated on GDMT before discharge.6 However, we still needed to know how fast to do it, how intense to be and the sequence of initiation to use.
The STRONG-HF study was a randomised, open-label, prospective clinical trial that helped to answer some of these questions.60 In STRONG-HF, patients admitted for an episode of decompensated HF who were not treated with full doses of GDMT were randomised, irrespective of LVEF, to a strategy of intensive treatment that consisted of initiation of a β-blocker, ACEI/ARB, or ARNI and an MRA at half doses before discharge and up-titrated to full doses within 2 weeks of discharge versus usual care (treatment according to local practices).60 In the high-intensity group, patients were monitored closely at 1, 2, 3, and 6 weeks after randomisation and assessed clinically and biochemically, including signs and symptoms of congestion, NT-proBNP, sodium, potassium, glucose, kidney function and haemoglobin. The study was stopped prematurely at the recommendation of the data and safety monitoring board because of a greater-than-expected difference in the primary endpoint between groups, favouring the intervention group.60 The primary endpoint (HF readmission or all-cause death) occurred in 15.2% of patients in the high-intensity group compared with 23.3% in the usual care group, with a risk difference of 8.1% (95% CI [2.9–13.2]; p=0.0021) and a risk ratio of 0.66 (95% CI [0.50–0.86]). At day 90, a greater proportion of patients in the high-intensity group were on full doses of medications (49% versus 4% for β-blockers, 55% versus 2% for RAASIs and 84% versus 46% for MRAs).60 BP and heart rate were also lower in the high-intensity group, and there was a greater improvement in congestion signs achieved with lower total daily doses of oral loop diuretics. The reduction in NT-proBNP was also significantly greater in the intervention arm, along with a more marked improvement in quality of life measured using the EQ-5D visual analogue scale. There were more adverse events at day 90 in the high-intensity arm than in the usual care arm (41% versus 29%), but most were mild and included cardiac failure (15% versus 14%), hypotension (5% versus <1%), hyperkalaemia (3% versus 0%) and renal impairment (3% versus <1%); the incidence of serious adverse events and fatal adverse events did not differ significantly between groups.60 The intervention of high-intensity care seemed to be equally effective and safe for older patients, for both men and women and irrespective of baseline NT-proBNP levels.61–63
Based on the data from STRONG-HF, the 2023 focused update of the 2021 European Society of Cardiology guidelines for the diagnosis and treatment of acute and chronic HF gave a class I level b recommendation for an intensive strategy of initiation and rapid up-titration of evidence-based treatment before discharge and during frequent and careful follow-up visits in the first 6 weeks following an HFh to reduce the risk of rehospitalisation or death.64 During the conduct of the trial, SGLT2Is were either not approved or not widely used; therefore, only 10% in the high-intensity group and 5% in the usual care group received them; however, the 2023 guidelines recommend including an SGLT2I as a component of the STRONG-HF strategy.60,64 Even though the STRONG-HF strategy was proved in patients with acute decompensated HF, the strategy could easily be used in outpatients with HF, provided there is adequate follow-up with clinical and biochemical assessment.
There are no head-to-head randomised control trials comparing different strategies of GDMT initiation and up-titration in patients with HF. Shen et al. modelled different approaches for initiation and up-titration of the different components of the foundational therapy for HFrEF using combined data from the placebo arm of the SOLVD-T and CHARM-Alternative trials that served as a ‘treatment-naïve’ population and assessed the impact of sequentially adding five therapies in a composite of cardiovascular death or HFh and all-cause death using data from pivotal trials conducted in HFrEF patients to test β-blockers, ACEIs, MRAs, ARNIs and SGLT2Is.65 In their model, the conventional sequence of starting an ACEI followed by a β-blocker, then an MRA, then switching the ACEI for an ARNI, and finally adding an SGLT2I took the longest to complete up-titration and had the lowest impact on the composite endpoint. In comparison, a sequence that involved first initiating an SGLT2I followed by an MRA, next an ARNI and finally a β-blocker (or a β-blocker followed by an ARNI) reduced by half the time taken to complete up-titration and doubled the effect on the endpoint; the effect was further enhanced by combining SGLT2I and MRA as the first step.65 In light of these data, we could say that the best thing we can do for our patients with HF is to abandon the conventional treatment sequence and try to initiate with a faster and stronger strategy, as shown in the STRONG-HF trial for most of our patients and – for those in whom this is not possible – to try to implement a strategy that allows for a fast implementation of the complete therapy.60,65 In this setting, initiating an SGLT2I that does not require up-titration plus an MRA that requires only two steps of titration and has a shorter time to clinical benefit, with reassessment in 1 week to evaluate tolerance and with attempts to add the remaining medications in the next 2–3 weeks, could be a reasonable option (Figure 3).9,10,52,65
Tailored Treatment for Heart Failure
Not every patient with HF is the same, and there are typical patient profiles that depend mostly on heart rate, BP, the presence of AF and CKD.51 Depending on the presence or absence of these variables, some patients with HF might not be eligible for some therapies and might need a personalised treatment regimen using other therapies that have also proved to be effective, such as ivabradine, vericiguat, and others, to try to give our patients the maximal benefit.66,67 Although in-depth discussion of such patient profiles and the available options for treatment is beyond the scope of this review, some detailed reviews and position statements are available.51
Conclusion
Treatment for HF has been revolutionised in recent years, increasing the benefit for patients by improving major outcomes and quality of life. Unfortunately, implementation of these new therapies is still far from optimal, which might be because of clinical/therapeutic inertia, along with misconceptions and unfounded fears around using HF medications. New data from randomised clinical trials show that rapid implementation of HF therapies is effective and safe, which should help us overcome the fear of treating HF patients. In light of these data, we could say that the best sequence/strategy of initiation and up-titration of GDMT is the one that best fits our patients’ needs. Clinicians should understand that successful implementation of GDMT relies on close follow-up of patients after initiation of treatment to evaluate tolerance, detect and correct adverse effects and to up-titrate medications to maximal doses. For most patients, the initiation of four medications at half-doses with up-titration to full doses in 2 weeks seems reasonable. For patients in whom tolerance or adverse events are a concern (BP on the lower limit, potassium on the higher limit) initiating two medications first, with rapid follow-up for reassessment of tolerance and initiation of the rest of the treatment might be a reasonable strategy. Consideration of BP, heart rate, kidney function/hyperkalaemia and the presence of AF allows us to provide tailored treatment based on different HF phenotypes. All these considerations might help us to offer a better, faster and stronger therapy to our HF patients. 