










Certain patients with cardiac amyloidosis (CA) and advanced heart failure may be considered for heart transplantation. However, extra-cardiac amyloidosis can significantly diminish the functional and survival benefits of such advanced therapies. As transplant teams carefully assess the severity and prognosis of organ-specific disease in heart transplant candidates, they must also consider systemic processes such as frailty and malnutrition.
As we review outcomes associated with extra-cardiac amyloidosis, we present a granular approach to identifying patients with CA and advanced heart failure who may benefit from heart transplantation.
Rise in Transplant Candidates
Survival outcomes in patients with transthyretin (ATTR) and light chain (AL) CA are influenced by not only progressive heart failure but also extra-cardiac disease.1,2 This natural history guides patients’ candidacy for advanced strategies such as heart transplantation.
Modern therapies for both ATTR and AL amyloidosis have improved patient survival and functional status, potentially rendering heart transplantation feasible for more CA patients with advanced heart failure. Examples of such drugs include ATTR stabilisers such as tafamidis, ATTR silencers including small interfering RNAs such as patisiran and vutrisiran, antisense oligonucleotide inotersen and AL therapies such as bortezomib and daratumumab.3–10
Several mechanisms may contribute to progressive cardiomyopathy despite targeted precursor protein treatment. For example, among wild-type and variant ATTR amyloidosis patients treated with tafamidis in the ATTR-ACT trial and the subsequent open-label extension study, over a median follow-up of 58.5 months, heart failure worsened in 2% and 10%, respectively, requiring heart transplantation, and mortality rates were 38% and 45%, respectively.3
In vitro examinations of neural cells and cardiomyocytes have demonstrated that ATTR monomers, ATTR oligomers and light chains are cytotoxic. ATTR silencer therapies produce an 85% reduction in circulating TTR; the remaining misfolded protein can, conceivably, continue to be deposited in patients with variant ATTR. In addition, wild-type ATTR can continue to accumulate in variant amyloid deposits, as has been demonstrated in patients with variant ATTR who underwent liver transplantation.4,11–15
In the modern context of effective therapies that reduce precursor protein production, heart transplantation is an important strategy for certain patients with progressive cardiac disease. Indeed, the proportion of heart transplants in the US performed for CA has increased from 0.3% of all heart transplants between 1987 and 2007 to 1.2% between 2008 and 2013 and 1.7% between 2010 and 2019.16,17
Extra-cardiac Manifestations and Severity
Evidence of Extra-cardiac Manifestations
As patients with ATTR or AL CA are considered for heart transplantation, transplant team members must consider that extra-cardiac disease can adversely affect outcomes after heart transplantation. Amyloidosis can affect multiple organ systems, including the nervous system, the gastrointestinal (GI) tract and liver, the respiratory tract and the kidneys (Table 1).18
However, while amyloid deposition can be associated with symptoms, the extent of deposition does not correlate perfectly with disease severity. In addition to amyloid deposition, toxicity from transthyretin fibrils or light chains may contribute to organ dysfunction. Finally, the distribution of organ involvement may vary depending on amyloid subtype.
In earlier reports of heart transplantation in CA patients with advanced heart failure, extra-cardiac disease at the time of transplant was described through clinical assessment, tissue diagnosis at transplant and imaging.
For example, among 11 AL CA patients undergoing heart transplantation between 1994 and 2005, one patient was noted to have peripheral neuropathy, macroglossia and GI involvement, and another had renal involvement identified at transplantation.19
In a UK series of CA patients undergoing heart transplantation between 1982 and 2002, extra-cardiac involvement was assessed using serum amyloid P component scintigraphy. Six of seven non-AL patients underwent imaging: one with ApoA1 amyloidosis had liver, kidney and splenic involvement; and another with variant ATTR had renal involvement. Of 13 out of 17 AL patients similarly assessed, 10 had renal, splenic, hepatic and/or bone marrow involvement.20
Renal insufficiency, as defined by proteinuria, renal biopsy or estimated glomerular filtration rate (eGFR), has been reported in recent case series of AL CA patients selected for heart transplantation. Comparing the transplant eras of 2002–2007 and 2008–2017, one centre demonstrated that, while patients selected for heart transplant had proteinuria, the average degree of this decreased from 0.28 g/day (25th–75th percentile 0.13–1.43 g/day; n=16) to 0.10 g/day (0.10–0.20 g/day; n=16) between these periods.21
At another transplant centre, two of 13 AL CA patients transplanted between 2004 and 2017 were found to have renal involvement by biopsy and neither had proteinuria ≥1 g/day. One of these recipients had progressive renal failure and required dialysis seven years after a heart transplant.22
In 13 AL amyloidosis patients transplanted at a third centre between 2010 and 2018, the average eGFR before transplant was 47.7 ± 27.6 ml/min/1.73 m2 and improved to 60 ± 61.2 22 ml/min/1.73 m2 over the 3-year follow-up period. Notably, one-third of patients underwent simultaneous heart and kidney transplantation and none had proteinuria ≥500 mg/day before transplant. The need for dialysis after heart transplantation was not reported.23
Neuropathy was reported in 77% of AL CA patients undergoing transplant at one centre, but categorisation by autonomic and peripheral systems was not available.23
Another analysis reported autonomic neuropathy in one of 13 AL CA patients and one of 18 ATTR CA patients undergoing heart transplant, and peripheral neuropathy in six of 18 ATTR CA patients undergoing heart transplant.22
Lastly, amyloid deposition in the oesophagus or stomach was reported in 11 of 13 AL CA and one of 18 ATTR CA patients transplanted between 2004 and 2017; deposition in the colon was reported in all 13 AL CA and one of 18 ATTR CA patients in the same series.22
Impact of Extra-cardiac Manifestations
In earlier eras of heart transplantation, extra-cardiac manifestations were associated with poor survival. United Network of Organ Sharing analyses of heart transplantation in cardiac amyloidosis over 1987–2010 (n=142) and 1987–2002 (n=69) demonstrated 1- and 5-year survival rates of 75–79% and 47%, respectively.24,25
However, larger registry analyses do not provide further comparisons by amyloid subtype, degree or type of extra-cardiac involvement, or cause of death. Older, smaller case series describe mode of death and the contribution of extra-cardiac disease.
For example, among 24 amyloidosis patients undergoing heart transplantation in the UK over 1982–2002, survival at 1 and 5 years after a heart transplant ranged between 50% and 20% in AL patients who did not receive chemotherapy; 71% and 36% in AL patients who did receive chemotherapy; and 86% and 64% in non-AL patients. Seven of the 10 deaths in the AL cohort were attributed to gastrointestinal, pulmonary or renal amyloidosis, while neither of the two non-AL patient deaths were due to amyloidosis.20
Extra-cardiac Manifestations and Outcomes in AL Amyloidosis
Light-chain control is key to improving survival outcomes after heart transplantation in AL amyloidosis recipients and has improved in the modern era of AL therapies.
In an older series of 11 patients who underwent heart transplantation and stem cell transplant (SCT) but no routine maintenance chemotherapy between 1994 and 2005, Lacy et al. reported that nine had >50% decrease in serum immunoglobulin free light chains, but only four remained in continued response during follow-up.19 Three died from progressive amyloidosis, one had a renal relapse and two died from SCT-related mortality, including infection, renal and hepatic failure.19
Recent single-centre case series of highly selected patients have reported robust longer-term light chain control after heart transplant, using strategies such as proteasome inhibitors, daratumumab and SCT. For example, in their series describing 13 AL amyloidosis patients transplanted between 2004 and 2017, Barrett et al. reported no cases of relapse, with five patients undergoing SCT and six requiring therapies such as proteasome inhibitors or daratumumab. No patients died of amyloidosis.22

In a more recent series of 13 AL amyloidosis heart transplant recipients between 2010 and 2018, six patients underwent SCT after heart transplant, with four of them requiring chemotherapy because they had a relapse. None of the four deaths were attributed to amyloidosis.23
Extra-cardiac Manifestations and Outcomes in ATTR Amyloidosis
TTR stabilisers and silencers will likely modify the natural history of progressive ATTR amyloidosis after heart transplant, but little data have been reported in the heart transplant population to date.23,26
Certainly, as heart transplantation improves survival in ATTR patients with CA, transplant teams will increasingly encounter extra-cardiac manifestations of amyloidosis. For example, an older report of seven wild-type ATTR cardiac amyloidosis patients undergoing heart transplantation between 2007 and 2015 did not report the use of ATTR-specific therapies, but only two of these patients remained free of extra-cardiac disease, such as dysmotility, autonomic or peripheral neuropathy, or carpal tunnel syndrome, after transplant.27
Another analysis of 12 patients with wild-type and variant ATTR cardiac amyloidosis describes amyloidosis progression after heart transplantation between 2002 and 2019.26 Before transplant, five patients had amyloidosis causing carpal tunnel syndrome and/or peripheral neuropathy. At a median of 4 years after transplant, eight patients had lumbar spinal stenosis, carpal tunnel syndrome, neuropathy and/or biopsy-proven GI amyloidosis. Severity of symptoms was assessed only after transplant, using the self-reported Composite Autonomic Symptom Score (COMPASS-31) and a simple clinician staging system, the polyneuropathy disability score (PND), which categorises patients by neuropathy and ambulatory ability.28 Two of the eight patients were on tafamidis and one on patisiran. At this centre, of an additional eight patients without amyloidosis surveillance who died after heart transplant, five died because of progressive GI amyloidosis or neuropathy. Two of the five had neuropathy or GI symptoms before transplant.
Earlier case series of CA heart transplants in patients with variant ATTR over 1982–2002 (n=3) and 2002–11 (n=9) highlight the use of either simultaneous or sequential liver transplantation in 100% and 66% of patients, respectively, as an attempt to reduce further amyloid deposition and progressive disease including neuropathy.20,26
However, after liver transplant, wild-type TTR can continue to accumulate in variant amyloid deposits, leading to progressive peripheral neuropathy and autonomic dysfunction among other problems.15,29,30
Moreover, in the modern era, therapies such as TTR silencers can significantly reduce circulating TTR without the additional morbidity from having a dual organ transplant. Recent reports of heart transplantation in patients with variant ATTR have described the use of heart-liver transplant infrequently, and this has primarily been in patients with concurrent cardiac cirrhosis.22,26,23
Extra-cardiac Disease, Downstream Effects and Patient Selection
Malnutrition and Frailty
Neuropathy and GI involvement in ATTR and AL amyloidosis can, conceivably, lead to a high prevalence of malnutrition and frailty in patients with CA. As in the overall population of patients referred for heart transplantation, these markers may prove useful as predictors of outcome in CA for those being considered for transplantation. 31,32
Among patients with amyloidosis, nutrition and frailty have been quantified using symptom assessments and scores based on laboratory values and weight. For example, among patients with AL CA, dysphagia occurs in 6–25%, diarrhoea in 29% and early satiety in 23%. Weight loss of 9 kg (20 lb) or greater is reported in 17–70% of patients.33
As measured using modified BMI, malnutrition occurs in up to 64% of variant ATTR cardiac amyloidosis patients and 59% of those with wild-type ATTR cardiac amyloidosis.34,35 Frailty is estimated to occur in 33–50% of patients with wild-type ATTR cardiac amyloidosis, in domains including (among others) autonomy, balance and muscle weakness.36
Such estimates are generally comparable to the prevalence of moderate-to-severe malnutrition and frailty among all patients evaluated for heart transplantation, but their prevalence after heart transplantation in cardiac amyloidosis has been reported only occasionally.23,32,31 Their effect on survival in heart transplant recipients with amyloidosis has not been ascertained.16,22,23,26
Organ-specific Selection Criteria
Organ-specific selection criteria for heart transplantation in patients with cardiac amyloidosis have been generally described in consensus guidelines and single-centre reports as part of efforts to improve post-transplant outcomes in this population in the modern era, but precise, acceptable thresholds for extra-cardiac disease severity and symptoms differ between centres.19,22,23,37 There are several reasons for this variation.
Transplant centres may differ in the extent of resources they can devote to post-transplant amyloidosis management, which requires close collaboration between multiple medical specialists and pharmacists.38 In addition, programmatic risk tolerance will vary depending on the consensus established at each centre among treating cardiologists, haematologists, nephrologists and neurologists.
As an example, for patients with AL amyloidosis, a recent American Society of Transplantation consensus statement proposed that in collaboration with oncologists, solid organ transplant physicians consider only candidates with no high-risk cytogenetics, good functional status with single organ involvement, a robust light-chain response to therapy, haematologic remission for >6 months and who are good candidates for eventual stem cell transplant.39
However, recent single-centre case series variably rely on only some of these factors in deciding candidacy for heart transplantation and, in the modern era of improved chemotherapy and immunotherapy, it is unclear that stem cell transplantation eligibility, in particular, should be a criterion.22,23
Transplant centres also vary in their patient selection criteria for simultaneous heart and kidney transplants in AL amyloidosis patients. Renal insufficiency in this population extends from proteinuria to anuria and dialysis dependence.40
Among patients with renal AL amyloidosis, severe proteinuria (defined either as >5 g/day and eGFR<50 ml/min/1.732 or as 24 hours’ protein: eGFR ratio <30 mg/ml/min/1.73m2) is associated with a significant risk of progression to dialysis dependence.41,42 The risks of acute and chronic nephrotoxicity associated with immunosuppressive regimens after heart transplantation alone may be perceived as prohibitive for heart-kidney transplantation in such patients, but individual centre volumes are too small to accurately describe post-transplant outcomes. 43,44
For AL amyloidosis patients with end-stage renal disease, 65% of US renal transplant clinicians recently surveyed reported a lack of consensus on which patients were appropriate for renal transplant, with the majority expressing concern about long-term survival.45
Common Principles for Organ System Evaluation
Patient selection for heart transplant must be informed by close discussion and coordination among multiple specialties at the transplanting centre, both before and after transplant. Importantly, specific extra-cardiac, organ-system-based assessments should be accompanied by a thorough appraisal of nutritional status and frailty.
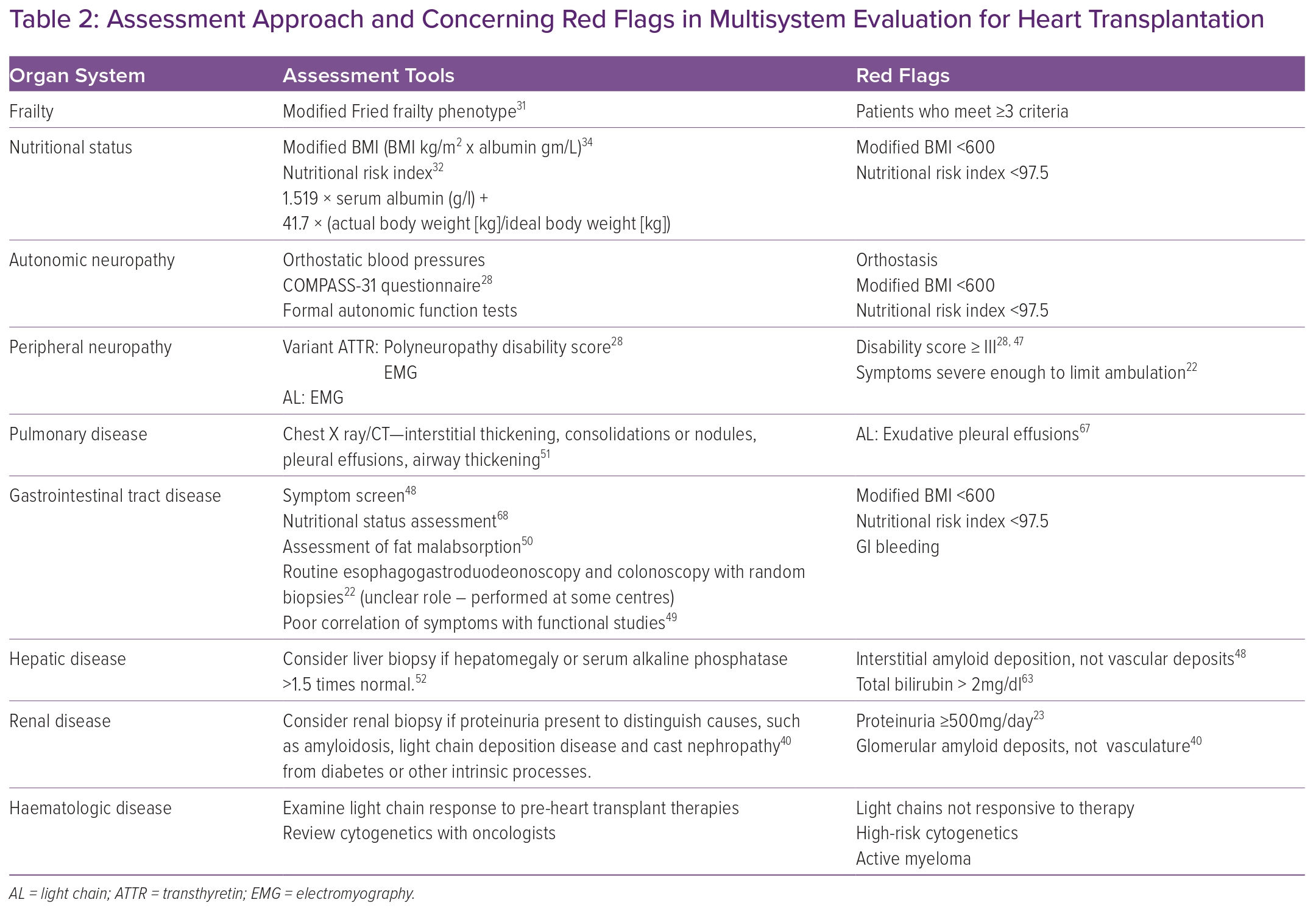
Relying on prognostic markers assessed in larger populations of heart transplant candidates and in patients with amyloidosis, we propose assessment approaches and red-flag benchmarks for organ-specific disease; adequate response to light-chain suppressive therapy in AL amyloidosis; and systemic markers, including functional and nutritional status in CA patients being considered for heart transplantation (Table 2).31,32,34,46,47
In many organ systems, amyloid deposition alone is not a contraindication to transplantation, as it does not predict dysfunction and poor outcomes after heart transplantation. This is particularly true for amyloidosis of the GI tract. Symptoms and nutritional status do not correlate perfectly with amyloid deposition, as autonomic neuropathy can also lead to diarrhoea, constipation, weight loss and early satiety.48 Therefore, the role of routine gastrointestinal tract biopsies is not clear, although they are performed at some centres.22 While certain functional studies, such as gastric emptying, do not correlate well with symptoms, a thorough symptom screen and objective measures of fat malabsorption may prove more useful.48–50
Assessing patient-reported symptoms in addition to certain functional measurements similarly facilitates the evaluation of amyloidosis-related peripheral and autonomic neuropathy. Such a strategy may include patient-completed questionnaires such as COMPASS-31, provider assessments such as PND and objective measures such as electromyography.28
While neurologic and GI involvement can be seen in both AL and ATTR amyloidosis, certain other organ systems are affected only in AL amyloidosis, for example, the pulmonary, hepatic and renal systems (Table 1). In such patients, invasive testing such as tissue biopsy can be considered if screening chest imaging or laboratory values such as alkaline phosphatase are abnormal.51,52
Renal biopsy should be considered in AL CA patients with proteinuria, particularly to distinguish between renal amyloidosis or light chain disease and other processes such as diabetes.40 In these patients, assessment of renal function using serum creatinine and eGFR may be challenging because of malnutrition; however, alternative measures such as cystatin C clearance have not been studied in this population. 53
Transplant programmes should couple these systematic evaluations with mechanisms to track decision outcomes in all CA patients referred for heart transplantation. Not all transplant centres may be able to offer advanced therapies to this population based on programmatic risk tolerance. Therefore, data on patient evaluation and decision-making, collected in multi-centre registries, may delineate geographic and socioeconomic disparities in access to heart transplantation for CA patients.
Conclusion
Outcomes for heart transplantation in cardiac amyloidosis have improved in the recent era of transplantation, due in part to successful disease-modifying therapies and more stringent patient selection.
At the time of evaluation for heart transplantation, teams must pay specific attention to the severity and natural history of extra-cardiac disease. We propose a systematic organ-based approach, with a particular focus on nutrition and frailty, to identify patients who could most benefit from heart transplantation in terms of functional and survival outcomes.
At individual transplant centres, tracking patient-specific data and decision outcomes in cardiac amyloidosis will help the transplant community ascertain the prevalence and severity of extra-cardiac amyloidosis in patients with advanced heart failure.