









The increasing progress in cancer therapies has reduced mortality rates for many cancers. Unfortunately, many life-saving therapies are burdened by the risk of cardiotoxicity (CTX). The cardiovascular system appears to be particularly susceptible to the action of many anti-neoplastic drugs, which may cause vasospastic or thromboembolic ischaemia, arterial hypertension, dysrhythmia, and left ventricular (LV) dysfunction, leading to heart failure (HF).1–7 These problems are even more relevant in an ageing population as cancer can occur in patients with pre-existing cardiovascular conditions.8
Some of these side-effects may occur or persist once the cancer is eliminated or controlled. Asymptomatic reduction in LV function and HF are the typical complications of cancer therapies in the long term.9 Many studies have tried to clarify the mechanisms underlying cancer therapy-related HF.10
Anthracyclines (ANTs) are the most studied cardiotoxic drugs. The main mechanism hypothesised for their cardiotoxicity is direct damage of cardiomyocytes through the production of reactive oxygen species (ROS) and reactive nitrogen species.11–15 With the increasing use of new biological drugs, other mechanisms of CTX have been observed, with drugs that affect the heart through secondary mechanisms.16 Newer intracellular signalling inhibitors block pathways of primary importance for myocardial function, especially under conditions of cardiac stress, such as hypertension or hypertrophy.17 Furthermore, in recent years, a growing incidence of myocarditis, due to the use of immune checkpoint inhibitors that unleash immune responses, has been recorded.18–21
This situation is complicated by the fact that novel biological drugs are sometimes combined (concomitantly or sequentially) with traditional chemotherapies. A typical example is the anti-ErbB2 receptor antibody trastuzumab, which can lead to LV dysfunction on its own and in people without pre-existing cardiovascular disease, but also unmask or worsen LV dysfunction in patients previously treated with ANTs, by interfering with the neuregulin/ErbB2 pathway that seems to modulate the increase in ROS-caused ANTs.1,2,22,23
Anthracyclines
ANTs are a typical example of cardiotoxic anticancer drugs, and their effects have been observed and studied since the 1960s.24 ANT-induced cardiomyopathy is characterised by the occurrence of cardiomyocyte damage that can eventually lead to HF. ANTs are a keystone in the treatment of many cancers, such as lymphomas, leukaemias and sarcomas, but also for early or advanced breast cancer.17 Their side-effects are usually dose-dependent and more frequently detected in the first year after completing treatment.25,26 ANT CTX can manifest acutely in up to 30% of patients soon after infusion, requiring either modification or withdrawal of anticancer regimens. Risk factors have been identified for the development of cancer therapy-related HF, pre-existing heart disease and advanced age.25 Moreover, there seems to be a gender-related predisposition. Although experimental data point towards better resistance from women regarding cardiotoxicity with involvement of mitochondria and less oxidative stress, very few studies have been conducted in humans, although female patients in clinical studies appear to be more susceptible to doxorubicin-induced cardiotoxicity.2,27,28 This apparent paradox may be explained because both age and menopausal status seem to be the two most important determinants of the sex-specific differences observed in the clinical setting, with higher susceptibility in prepuberal girls and post-menopausal women. Studies in young children receiving anticancer drugs for haematological malignancies suggest that prepuberal girls are more susceptible to develop early or late cardiac toxicity than boys of the same age.27,29 These data are consistent with the absence of female hormones at this age. Unfortunately, no survey has been conducted to specifically assess sex differences in the occurrence of ANT cardiotoxicity in adults.
Several mechanisms underlying anthracycline CTX have been observed, but the main ones we will focus on are induction of oxidative stress, activation of DNA damage responses and impairment of mitochondrial biogenesis and metabolism. The consequence of these processes is cardiomyocyte death, apoptosis and necrosis, while the surviving cardiomyocytes develop maladaptive changes. This leads to pathological remodelling of the LV, with dilatation and impairment of contractility, until the decline of systolic function and development of clinically manifest HF.9
ANTs are characterised by a marked susceptibility to be rapidly reduced to unstable metabolites, such as doxorubicin-semiquinone, which generate hydrogen peroxide and superoxide by reacting with oxygen. ROS are also produced thanks to the ability of these drugs to chelate intracellular free iron, creating iron-doxorubicin complexes that react with oxygen (Figure 1). Furthermore, ANTs can directly interfere with the activity of major iron-transporting and iron-binding proteins, such as ABCB8, a mitochondrial iron exporter, promoting mitochondrial iron accumulation and ROS production. It has also been observed that hearts from patients with doxorubicin-related heart dysfunction have significantly higher mitochondrial iron levels than in patients with other types of cardiomyopathies or normal cardiac function.30
Research led by Edward Yeh has shown that the production of ROS could also be secondary to the interaction of ANTs with the beta isozyme of topoisomerase 2 (Top2), the only isoform expressed by adult mammalian cardiomyocytes.31 While the interaction of the drug with Top2-alpha – overexpressed in proliferating cancerous cells but not in quiescent tissues – generates a ternary Top2-doxorubicin-DNA cleavage complex that in turn triggers the death of tumour cells, the Top2-beta-doxorubicin-DNA complex induces DNA double strand breaks, ultimately promoting cardiomyocyte death.31 The ensuing DNA break induces the activation of p53, an enzyme that activates the proteins responsible for the DNA repair process, but can also repress genes involved in mitochondrial biogenesis, such as PPARGC1, oxidative phosphorylation, ultimately leading to defective organelle biogenesis and metabolic failure.31 An abnormal accumulation of mitochondria damaged by doxorubicin in the myocardium has also been reported, promoting the production of ROS and the death of cardiomyocytes. This accumulation would seem to be caused by the activation of p53, which is able to inhibit the normal recycling of dysfunctional mitochondria via autophagy.9
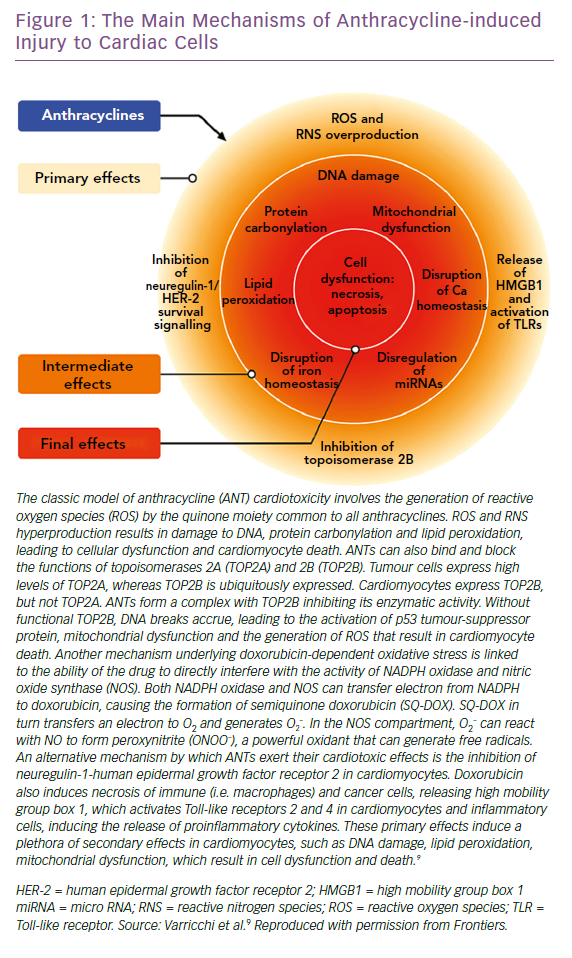
ANTs are also involved in the activation of the mitogen-activated protein kinase (MAPK) cascade through reactive oxygen species and Ca2+. In particular, it is worth mentioning the role of p38 MAPK in the induction of cardiomyocyte death.32 It has been demonstrated that before any clinical sign of LV dysfunction in ANT cardiotoxicity, there is a reduction in the phosphocreatine:adenosine triphosphate ratio, suggesting the presence of alterations in myocardial energetics.33 In this study, the authors also demonstrated that ANTs can affect the normal functioning of creatine kinase (CK) by oxidising its sulfhydryl groups. More studies on this pathway are needed to identify novel cardioprotective therapeutic approaches. The possible protective role of CK in heart diseases is supported by improved cardiac function in murine hearts overexpressing myofibrillar CK and subjected to pressure overload, compared with non-transgenic mice.34 Moreover, CK overexpression seems to improve cardiac function and general myocardial energetics and also the survival of mice affected by CTX induced by ANTs.35
Among their other effects on cardiomyocytes energy metabolism, ANTs can alter fatty acid oxidation, due to a reduction of the phosphorylation of the enzyme anti-acetyl-CoA carboxylase and of the intracellular concentration of 5’-activated protein kinase (AMPK). Further studies will be needed to clarify the role of AMPK in ANTs-induced HF.36,37
Several approaches have been proposed to reduce ANT CTX. Beside limiting the cumulative anthracycline doses, the interest of the scientific community has also been focusing on antioxidant drugs.2,3 However, none of these strategies is unanimously recommended, emphasising the need for further studies.8 The use of dexrazoxane has been clinically evaluated in children treated with doxorubicin for acute lymphoblastic leukaemia, resulting in reduced myocardial injury, as indicated by a decreased level of serum troponin T.15,38
Among traditional HF drugs, beta-blockers have been shown to reduce oxidative stress and calcium overload in myocardial cells.39,40 Carvedilol has been shown to have a preventive role against LV dysfunction in patients treated with ANTs reducing the production of ROS, apoptosis of cardiomyocytes and mitochondrial alterations.41–43 In some experimental models of ANT-induced cardiotoxicity, nebivolol was also able to improve LV function, increase nitric oxide (NO) levels and reduce oxidative stress.44,45 Nebivolol, used before ANT-based treatments, also reduced the incidence of LV dysfunction, compared with placebo.46
The renin-angiotensin-aldosterone system also plays a key role in ANT-induced CTX.47 In particular, in patients treated with ANTs, enalapril reduced the incidence of LV dysfunction when compared with placebo.48 In vitro and in vivo experiments demonstrated the cardioprotective effects of angiotensin receptor blockers – candesartan can reduce in vitro ANT cardiotoxicity, while telmisartan can blunt acute LV dysfunction induced by doxorubicin when administered pre- and post-chemotherapy in rats.49 Furthermore, telmisartan can inhibit the production of TNF-alpha and interleukin 6 and can affect the availability of NO.50 It also seems that the co-administration of angiotensin-converting enzyme inhibitors and carvedilol can reduce cardiac damage induced by ANTs.51
Biological Drugs
Anti-ErbB2 Drugs
ErbB2 (also known as HER-2/NEU) belongs to the epidermal growth factor receptor (EGFR) family. These receptors can homodimerise or heterodimerise and are phosphorylated upon binding with their ligands, initiating several cellular responses.52 ErbB2 is overexpressed in 25–30% of breast cancers and this has led to research specifically targeting drugs such as trastuzumab, pertuzumab and lapatinib.53
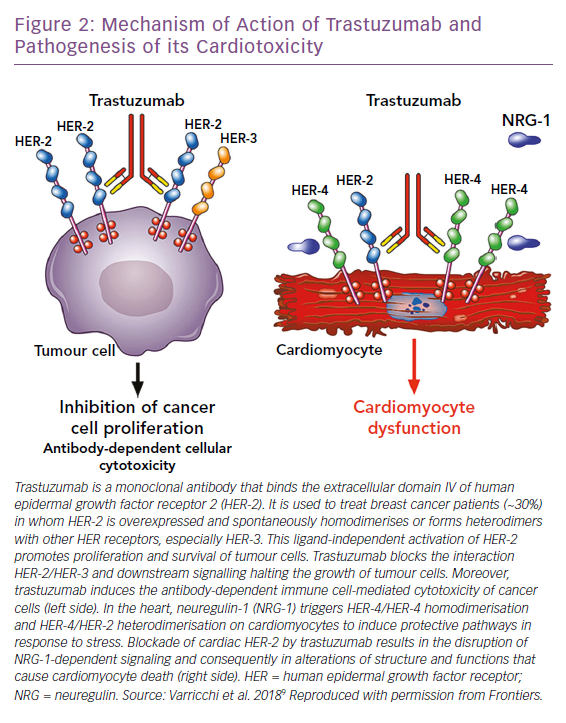
Trastuzumab is the prototypical biological drug. It is a humanised monoclonal antibody that targets ErbB2, binding to its extracellular domain IV, and has revolutionised ErbB2+ breast cancer protocols since its introduction in 1998. It can also cause CTX that spans from asymptomatic decreases in LV ejection fraction (LVEF) to congestive HF.1,10,54 Most patients with little or no risk factors can tolerate trastuzumab for long periods of time. Given the importance of this drug in ErbB2+ breast cancer, the Cardiac Safety Study in Patients With HER2 + Breast Cancer (SAFE-HEaRt study) has been designed to evaluate whether anti-HER2 therapies can be given to women with mildly reduced heart function and optimised cardiac therapy and monitoring.55
The mechanisms of CTX induced by ErbB2 blockers have not been fully elucidated (Figure 2). In the heart, neuregulin, secreted from endothelial cells, upon binding to ErbB4 induces the dimerisation of ErbB4 and ErbB2, thus activating protective trophic and pro-survival pathways in response to stress, such as hypertension, hypertrophy, or exposure to ANTs, and it has also been shown that it can modulate cardiomyocyte proliferation in mammalian hearts.9,12,56– 60 The inhibition of the neuregulin-1/Erbb2 axis weakens the myocardium and makes it vulnerable to myocardial injury. Timolati et al demonstrated a role of neuregulin-1 in the modulation of doxorubicin-induced oxidative damage, with an impact on antioxidant enzymes such as glutathione reductase, suggesting that trastuzumab may act as a modulator of ANT-related toxicity.23
The interactions between ANT and trastuzumab have been extensively studied. The coadministration of trastuzumab with ANTs in people with breast cancer, increased ANT toxicity in early trials and is now avoided.61–63 In fact, it has now been shown that anti-HER-2 drugs block the protective mechanisms of HER-2, exacerbating the oxidative damage caused by ANTs.12,64
ErbB2 knockout mice develop dilated cardiomyopathy and show a higher prevalence of cardiomyocyte death when treated with ANT.65 On the other hand, Belmonte et al. demonstrated that overexpression of ErbB2 in the heart reduced ROS levels, increasing the activity of glutathione peroxidase 1 and its co-activating factors such as c-Abl and Arg.66 The same group reported a bidirectional cross-regulation between ErbB2 and beta-adrenergic signalling pathways.67 Interestingly, patients treated with trastuzumab, ANTs, or both have been shown to be exposed to reduced risk of LV dysfunction when incidentally administered with beta-blockers.68 Recent data suggest that beta-blockers, such as bisoprolol and metoprolol are not able to fully prevent trastuzumab-induced cardiomyopathy, showing that blockade of beta-1 alone is not sufficient to protect the heart.69,70 While non-selective beta-blockers did not really prove beneficial in the ANT setting, these clinical and experimental findings support their use in the trastuzumab setting.67,71
Anti-vascular Endothelial Growth Factor Drugs
As seen above, ROS play a central role in the mechanisms of CTX induced by ANTs and by ErbB2 blockers. AMPK, which may have a role in ANT-induced cardiotoxicity, seems to be targeted also by the tyrosine kinase inhibitor sunitinib. Indeed, sunitinib is primarily known as a vascular endothelial growth factor (VEGF) inhibitor, but it is also a multiple tyrosine kinase inhibitor. Among many other kinases (>30), it can inhibit ribosomal S6 kinase, activating the intrinsic apoptotic pathway, and AMPK (usually activated by energetic stress), contributing to the reduction of adenosine triphosphate levels.52,72,73 Our preliminary findings show that CK can also modulate sunitinib actions on the contractile apparatus of cardiomyocytes by regulating oxidative stress.74,75
Additionally, it seems that sunitinib can prolong the opening time of the mitochondrial permeability transition pore, with consequent swelling and deformation of the mitochondria in murine cardiomyocytes affected by pressure overload.76 Conversely, studies have demonstrated that oxidative phosphorylation is not significantly affected by sunitinib and suggest that its CTX is less frequent than predicted.77
It has been shown that sunitinib damages pericytes and can affect the microvascular circulation of the heart, rather than impair cardiomyocyte functionality directly, and a recent paper has investigated the connection between afterload and sunitinib-induced CTX.78,79 Using a preclinical model of engineered cardiomyocytes (first murine and then human), Truitt et al. demonstrated that sunitinib can induce cardiomyocyte death, decrease the contractile force of the heart and generate spontaneous beating at clinical doses. They also found a correlation between an increase in the afterload and the CTX induced by sunitinib. According to these findings, antihypertensive therapies may be used to reduce the effects of sunitinib.79
Sorafenib is another tyrosine kinase inhibitor with significant CTX. Most of the information we have on CTX induced by sunitinib and sorafenib comes from two meta-analyses including almost 7,000 patients given sunitinib and 900 patients given sorafenib. These showed that 4.1% of patients treated with sunitinib developed HF, while 1% of patients treated with sorafenib had signs of cardiac dysfunction.80,81 It is important to highlight that both meta-analyses only included retrospective studies. So far, there are few data derived from prospective studies, although Schmidinger et al. demonstrated that three of 14 patients who had a cardiac event and were administered with sorafenib, developed LV dysfunction assessed by significant reduction of LVEF.82
Despite the aforementioned studies, the real incidence of CTX-induced by sorafenib is not yet clear and more studies are needed for this reason. Sorafenib can inhibit at least 15 different kinases, such as VEGFR, PDGFR, Raf-1/B-Raf, FLT3 and c-Kit.52,83,84 In addition, a 2018 study demonstrated that sorafenib has an intrinsic cardiotoxic effect on cardiomyocytes, impairing calcium homeostasis.85
Immunotherapy
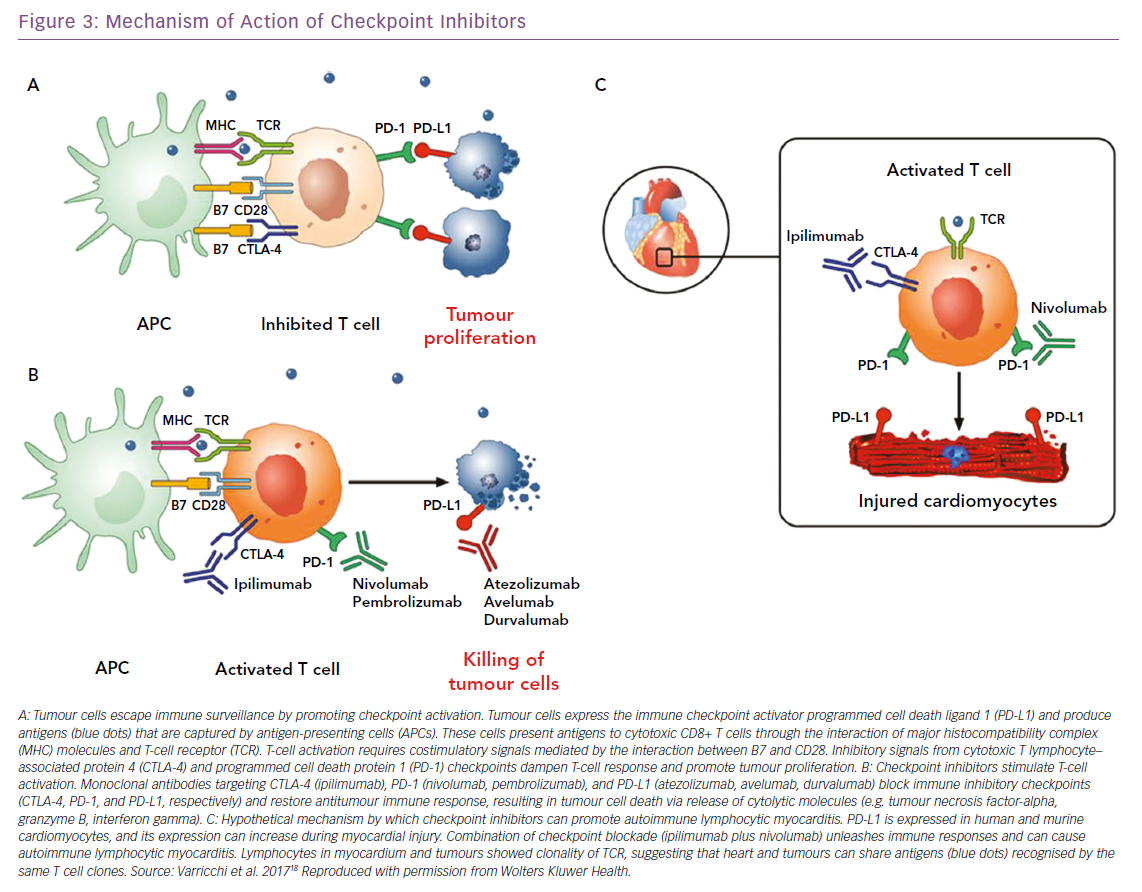
Over the past few years, cancer immunotherapies have revolutionised the clinical management of a wide spectrum of solid and haematopoietic malignancies. The forefront of immunotherapy is represented by immune checkpoint inhibitors (ICIs), whose purpose is to inhibit molecules such as cytotoxic-T-lymphocyte-associated antigen 4 (CTLA-4) and of programmed cell death 1 (PD-1) and its ligand PD-L1. CTLA-4, expressed on T cells, competes with CD28 in binding CD80 and/or CD86, expressed on antigen-presenting cells, modulating the amplitude of T-cell activation and showing immunosuppressive activity.86–88 This results in immunosuppression with downmodulation of T helper cell activity and enhancement of regulatory cells.
PD-1, expressed at low levels on T cells, activated natural killer cells, B cells, monocytes, immature Langerhans’ cells and cardiomyocytes, and its ligand PD-L1, constitutively expressed at low levels on both professional and non-professional antigen-presenting cells as well as on non-haematopoietic cells plays a fundamental role in the maintenance of peripheral tolerance and the prevention of autoimmune diseases.89 Monoclonal antibodies targeting CTLA-4 (ipilimumab), PD-1 (nivolumab, pembrolizumab), and PD-L1 (atezolizumab, avelumab, durvalumab) block these immune inhibitory checkpoints and restore the antitumour immune response, leading to tumour cell death through the release of cytolytic molecules, such as tumour necrosis factor-alpha, granzyme B and interferon-gamma (Figure 3).18 However, immune checkpoints play a central role in the maintenance of self-tolerance. Therefore, blocking these pathways can lead to imbalances in immunologic tolerance that results in immune-related adverse events.90 These side-effects are common, but fortunately in most cases they are reversible and not severe. They include mostly skin manifestations, such as pruritus, rash and vitiligo in 43–45% of patients, but also liver and gastrointestinal events that may occur 6–7 weeks after treatment was initiated. Greater concern is expressed on endocrinopathies, observed in about 6–8% of patients. They are the only immune-related adverse events with a high risk of irreversible toxicity and result from immune infiltration into either the thyroid or pituitary glands, causing thyroiditis or hypophysitis, respectively.91–93
When ICIs were introduced as cancer treatments, little attention was paid to cardiac side-effects. Then, isolated cases of fulminant myocarditis (Figure 3) and other cardiovascular disorders (pericarditis, vasculitis and AV blocks) were reported by several independent groups.19,94–97 The 2018 study by Mahmood et al. is significantly larger than previous reports.98 The authors present a retrospective, multicentre review of myocarditis in 35 patients and show that myocarditis presented early, with a median presentation of more than 30 days after starting ICIs, and 81% presenting within 3 months of treatment initiation. This suggests the importance of a surveillance protocol, especially in the initial phases of therapy when it may have the most impact. The same authors showed that serum troponin was abnormal in 94% of the cases, highlighting a possible role in early detection of ICI CTX. Instead, measurement of EF may be less useful for surveillance, because EF with myocarditis was normal in half of the cases. In fact, a preserved EF in not reassuring in ICI myocarditis, unlike other types of myocarditis where a normal EF is traditionally considered relatively benign and self-limiting.99
The development of myocarditis in patients treated with ICIs has a solid biological base. In 2001, a seminal paper by Nishimura et al. demonstrated that mice deficient for the CTLA-4 and PD-1 axes presented with autoimmune myocarditis and dilated cardiomyopathy, showing that these molecules can prevent autoimmunity.100 Furthermore, absence of PD-L1, or its inhibition, can worsen the survival from myocarditis, suggesting a role for PD-1/PD-L1 and CTLA-4 in limitation of T cell–mediated autoimmune myocarditis. Interestingly, PD-1 and PD-L1 were observed to be increased in cardiomyocytes from rat hearts subjected to ischaemia-reperfusion.100,101
Future Perspectives
Cardio-oncology is an ever-expanding field of research. In this article we have only discussed the studies conducted on ANTs anti-HER2 drugs and anti-VEGF drugs, but several other drugs (alkylating agents, antimetabolites, proteasome inhibitors, other, tyrosine kinase inhibitors, antimicrotubule agents) can generate LV dysfunction.17 A tight collaboration among cardiologists and oncologists is building up quickly.102,103
Immunotherapies have been introduced more recently, and in view of the fact that autoimmune myocarditis induced by ICIs has fulminant progression, including immunologists in this cardio-oncologic collaboration appears necessary for better management of ICI CTX.104 At the moment, beyond ICIs, novel monoclonal antibodies targeting several immune checkpoints, and new cancer therapies, such as engineered T cells, cancer vaccines and PI3K inhibitors are being studied and developed.105–108 A thorough cardio-immuno-oncologic collaboration seems fundamental, to the assessment of potential toxicities of current and novel drugs, in clinical as well as in basic research also considering that these drugs are often combined, thus increasing their cardiotoxic potential.99,109
In addition, novel data point to a direct relationship between cancer and the heart. Indeed, cancer and HF share common mechanisms, risk factors and comorbidities, while several studies have suggested that cancer cachexia can trigger cardiac dysfunction, and that cardiovascular health can predict all-cause mortality in cancer patients.8,110–117 More recently, experimental studies led by Rudolf de Boer have elegantly shown that HF stimulates tumour growth by circulating factors.118 Investigation of the mechanisms and pathways linking HF to cancer is a novel, but very promising field of research that aims to answer exciting questions of whether HF promotes malignancies.111