










Dyspnoea is one of the leading symptoms in patients presenting to the emergency department. It is estimated that more than 50% of hospitalised patients and up to one-third of ambulatory outpatient present – at least to some degree – with dyspnoea.1 Its prevalence increases with age and, in many patients, more than one aetiology of dyspnoea is present.2.
The incidence of pulmonary embolism (PE) and heart failure (HF) also increases with age and, not surprisingly, both conditions are among the most frequent causes of dyspnoea leading to hospitalisation, relevant morbidity and mortality.3
PE and acute HF not only have similar clinical presentations but also share many risk factors and pathophysiological mechanisms.4 For that reason, the diagnosis of coexisting PE and HF in dyspnoeic patients is challenging. The identification of one of these might lead to premature exclusion of the other and, as such, a diagnosis of concomitant PE and HF may be delayed, missed or not even considered by the treating physician.4 Furthermore, one condition may aggravate the other and the coexistence of both conditions has major therapeutic implications and detrimental effects on survival.5
In this paper, we summarise the pathophysiologic interactions of PE and HF, describe our diagnostic algorithm for PE in patients with HF and, based on the most recent recommendations on the treatment of PE and acute HF (AHF), we propose our therapeutic approach to patients with concomitant PE and AHF.5–8
Case Presentation
A 79-year-old woman with HF with preserved ejection fraction (HFpEF) presented to the emergency department reporting a sudden increase of breathlessness and chest discomfort over the last few hours. Arterial blood pressure was 160/95 mmHg, the pulse was regular with a heart rate of 115 BPM. The peripheral oxygen saturation (SpO2) was 88% by breathing ambient air (94% with 4 l/min of oxygen) and the respiratory rate 32/min. The physical examination was remarkable for pitting oedema on both legs, distended jugular veins and bilateral pulmonary crackles. Extremities were warm and appear well perfused. The ECG showed sinus rhythm; no repolarisation abnormalities were present. Levels for high-sensitivity troponin were mildly elevated without relevant changes after 1 hour. The patient was treated with loop diuretics for AHF without relevant improvement of symptoms. The junior physician on night shift duty suspected the presence of concomitant PE and called his senior to discuss how to further manage this patient.
Pathophysiology
Cardiovascular Disease and the Risk of Pulmonary Embolism
Arterial hypertension, dyslipidaemia, diabetes, obesity, tobacco use, unhealthy diet, stress and oestrogen therapy have major detrimental effects on endothelial function, inflammation and hypercoagulability, and may promote the occurrence of both atherothrombosis (leading to MI and HF) and venous thrombosis (leading to PE).9
A large registry study showed that previous MI significantly increases the risk of PE.10 The higher the burden of coronary artery disease, the higher the risk of experiencing PE.11 Patients with HF have a nearly doubled incidence and mortality of PE than those without HF – this risk increases as the cardiac function declines.12,13 A left ventricular ejection fraction (LVEF) <20% is independently associated with a 38-fold risk of a venous thromboembolism compared to patients without HF.14
Among patients with PE, patients with coexisting HF have less frequently identified triggers, such as recent surgery or active malignancy, and present less commonly with signs of deep venous thrombosis. Conversely, these patients are older and are more likely to have AF (32% versus 7%) and respiratory impairment (i.e. hypoxaemia and hypercapnia).13 Whether AF increases per se the risk of PE or whether it is merely a marker of comorbidities and severe cardiac disease remains a matter of debate.15,16 Since the prevalence of right atrial thrombi, potentially causing PE, in patients with AF has been described as low (<1%) compared to left atrial thrombi (9%), we question the clinical relevance of this association.17
PE Precipitating HF
PE may induce a rapid increase in pulmonary pressures and acutely precipitate HF by causing right ventricular dysfunction (acute cor pulmonale). The elevation of pulmonary pressure following PE is observed only when more than half of the pulmonary vasculature is obstructed by thrombotic material.18–20 This is because distension and recruitment of additional pulmonary capillaries might decrease vascular resistance and compensate for circulatory changes. When thrombotic occlusion extends to more than 50% of the lung vessels, pressure elevation occurs.
An unconditioned right ventricle (RV) can tolerate a mean pulmonary arterial pressure of up to ~40 mmHg. If the RV is exposed to higher pressures, it can either tolerate it (then an antecedent adaption of the RV secondary to a pre-existing elevation of pulmonary pressure must be assumed) or its function is impaired. The normal RV function is an interplay between preload, contractility, afterload, ventricular interdependence and heart rhythm.21
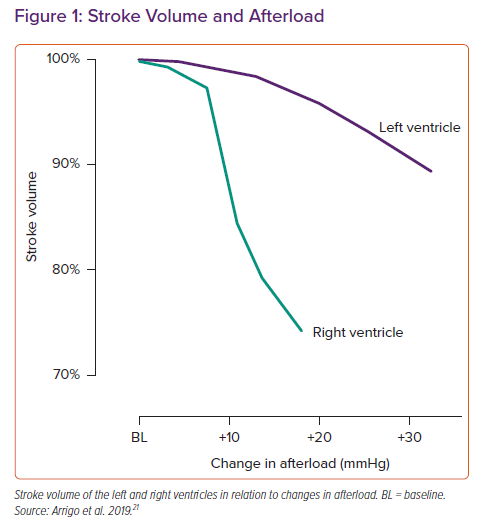
A massive and rapid increase in RV afterload as observed in the context of PE induces RV dilation and a reduction in RV contractility, leading to a drop in RV stroke volume (Figure 1), change in ventricular interdependence and an increase in systemic congestion.21 As a consequence of the reduced RV function and altered interdependence, left ventricular preload is impaired, which may cause a reduction in left ventricular stroke volume and hypotension. Increased RV wall tension, arterial hypotension and impaired oxygenation may precipitate RV ischaemia and arrhythmias, which further deteriorate cardiac function and cause profound haemodynamic instability and shock.5,22
In summary, PE and HF have similar clinical presentations and share many risk factors and pathophysiological mechanism. Patients with cardiac disease, such as coronary artery disease, AF and, in particular, HF, display a higher risk for PE. On the other hand, PE may precipitate RV and left ventricle (LV) dysfunction and induce AHF or shock.
Diagnosis
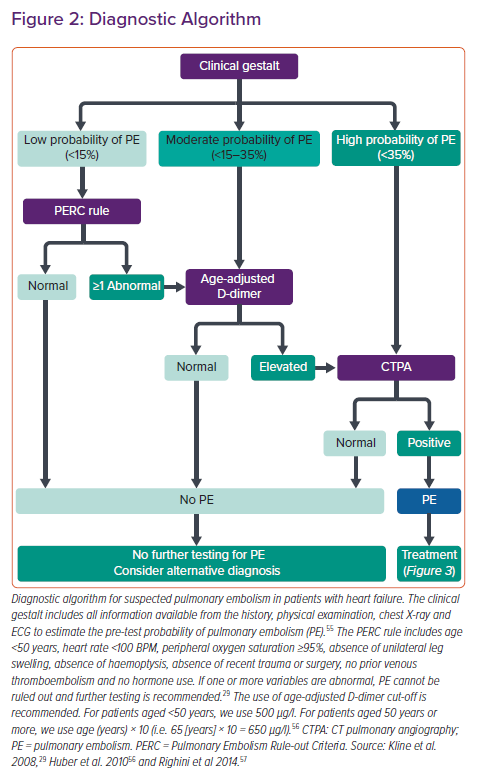
The diagnostic process for the presence of PE in patients with HF includes clinical gestalt, the use of designated scores, laboratory markers and CT pulmonary angiography (CTPA). Our own algorithm to approach this topic is summarised in Figure 2.
Clinical Gestalt, Scores and D-dimers
Every diagnostic process begins with the formulation of a pretest probability by integrating all clinical information obtained. In this context, it should be emphasised that the result of any medical test that was ordered to confirm or to rule out a given disease is useless if not seen in the light of pretest probability.23 For example, paroxysmal nocturnal dyspnoea, auscultation of a third heart sound and radiological evidence for pulmonary venous congestion with interstitial oedema might result in such a high suspicion for the presence of AHF that the additional information obtained by measurement of cardiac biomarkers (i.e. natriuretic peptides) lowers – not increases – the diagnostic yield.24,25
For the semi-quantitative estimation of pretest probabilities in patients with suspected PE, several scores have been developed, of which the Geneva and the Wells score are probably the most popular ones.26 These scores have proven to be useful tools to estimate probabilities for the presence of PE in a clinical setting. However, these tools are limited by the fact that both scores have been designed to lead to a further testing cascade, either by measurement of D-dimers (in cases of low pretest probability) or by CTPA (in cases of moderate or high probability). Moreover, the pretest probability for the presence of PE estimated by experienced clinicians is non-inferior or even superior to the probability calculated by one of these scoring systems.27,28 This difference might become more accentuated in the setting of a coexisting condition – such as AHF – for which no validated scores exist.
Therefore, in patients with signs and symptoms of AHF and suspected PE, we advocate to use clinical gestalt to estimate the pretest probability for PE. In this specific situation, congestive HF should be strongly considered as an additional persistent risk factor to develop venous thromboembolism. In case of a low pretest probability, the Pulmonary Embolism Rule-out Criteria (PERC) rule should then be employed as initial scoring system. The PERC rule, with a sensitivity >97%, allows to rule out PE without additional testing.29 Because there are many overlapping variables between specific scoring systems (Table 1), we discourage the use of the Wells or Geneva score followed by the PERC score.30
When the pretest probability for PE is estimated to be low and at least one PERC criterion is abnormal or when the pretest probability is moderate, D-dimers should be measured (Figure 2). In case of elevated age-adjusted D-dimers or when pretest probability is high, CTPA should be performed next (Figure 2). To our knowledge, no studies have examined the predictive value of scoring systems or have evaluated distinct cut-off levels of D-dimers in the context of PE and AHF. However, a negative PERC score or normal age-adapted levels of D-dimers appear to safely rule out PE even when accompanied by other conditions.29
CT Pulmonary Angiography
CTPA is, to date, arguably the gold standard to diagnose PE (European Society of Cardiology [ESC] guidelines class I indication). Here, we want to emphasise two points. First, according to Bayesian probabilities, in patients with high pretest probability for the presence of PE, up to 8% of chest imaging might yield false negative results.31 This might be particularly true in patients with acute right-sided HF caused by tumour microemboli, which, in contrast to paraneoplastic venous thromboembolism, might be too small to be detected by imaging methods. Hence, it remains controversial whether patients with a negative CTPA and a high clinical probability should be further investigated.6
Second, patients with acute right HF and subsequently elevated central venous pressure and venous congestion commonly experience worsening renal function. In fact, venous congestion, more than reduced cardiac output, is the best haemodynamic predictor for a reduced glomerular filtration rate.32,33 Conversely, however, the risk of inducing post-contrast acute kidney injury is overestimated.34 Acute contrast-induced nephropathy is defined as the iatrogenic worsening of kidney function following the administration of IV radiocontrast – it usually shows a mild course with spontaneous return to baseline renal function without long-term compromise. As such, in patients with moderate to high probability for PE, we advocate not withholding CTPA as one of the most important diagnostic cornerstones.
Cardiac Biomarkers and Echocardiography for Risk Stratification
The latest guideline paper from the American College of Chest Physicians proposes a pragmatic approach to the use of both echocardiography and cardiac biomarkers and, in particular, suggests not to use these diagnostic tools routinely in all patients with PE or in all patients with a non-low-risk profile.35
Cardiac biomarkers, such as high-sensitivity troponins and natriuretic peptides, are associated with higher risk of PE-related death but, since they are frequently elevated (in ~50–60% of patients with PE), they have a low positive predictive value and do usually not alter treatment decisions.6 In the clinical setting of concomitant acute PE and HF, the diagnostic yield of cardiac biomarkers is even further reduced. Because they have an excellent negative predictive value, natriuretic peptides and high-sensitivity troponins are of major use when found at normal levels. This, however, is an unlikely scenario in patients presenting with both PE and HF.
Routine echocardiography for the assessment of RV function is not recommended for the diagnostic workup of haemodynamically stable patients with PE.6 Indeed, echocardiographic evidence of RV dysfunction (e.g. RV:LV ratio ≥1.0, tricuspid annullar plane systolic excursion <16 mm) is common (prevalence of ~25% in unselected patients with PE) and associated with worse short-term outcome, but displays low positive predictive value for PE-related death.36
Conversely, in stable patients with concomitant (including acute) HF, echocardiography should be performed at admission or during the index hospitalisation to diagnose the underlying cardiac pathology that may require specific treatments and/or initiation of a neuro-humoral blockade (such as beta-blockers or renin–angiotensin–aldosterone system inhibitors).3,37
In patients with suspected or confirmed PE and haemodynamic instability, echocardiography should be immediately performed (ESC guidelines class I indication). Indeed, in unstable patients with suspected PE, the presence or absence of echocardiographic signs of acute cor pulmonale may rule in or rule out PE as a cause of haemodynamic instability and accelerate the diagnostic process and treatment delivery.6
Management
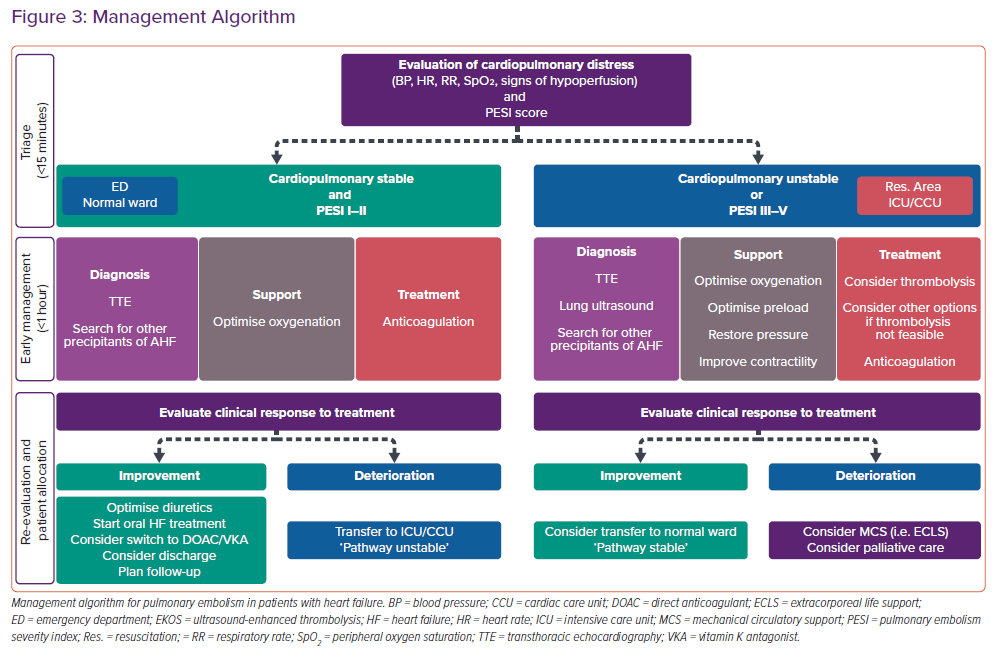
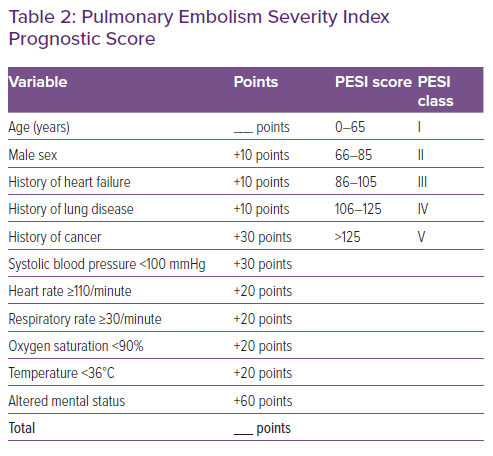
The management of patients with concomitant PE and HF/AHF may be challenging.5,21 The spectrum of disease may vary from oligo- or asymptomatic presentations to critical illness with profound cardiopulmonary instability. Therefore, the first minutes should be dedicated to triage, i.e. assessment of the cardiopulmonary distress based on the acquisition of the vital signs (blood pressure, heart rate, respiratory rate and peripheral oxygen saturation), the estimation of the perfusion state (search for signs of peripheral hypoperfusion, such as cold, mottled skin, altered mental state and oliguria), and the determination of the PESI score (Figure 3 and Table 2).5,21 Cardiac arrest, obstructive shock (systolic blood pressure <90 mmHg and signs of peripheral hypoperfusion), and persistent hypotension (systolic blood pressure <90 mmHg or requiring vasopressors) define haemodynamic instability.6
Low-risk Patients without Cardiopulmonary Instability
Low-risk patients presenting with cardiopulmonary stability and PESI I–II (Figure 3, left side) can be managed in a regular emergency department or a normal ward.
The management should consist of three parts delivered simultaneously. First, the diagnostic process should be refined by searching for other precipitating factors of AHF (e.g. myocardial ischaemia, arrhythmia, infection, uncontrolled hypertension and non-compliance) and performing a transthoracic echocardiogram (TTE) to better understand the underlying cardiac pathology leading to AHF and assess the presence of cardiac sequelae of PE (e.g. RV dysfunction and pulmonary hypertension).38,39 The additional information derived from the finalised diagnostic process may allow the optimisation of the treatment directly tailored to the underlying precipitating factor and cardiac pathology.40
Second, oxygenation should be optimised by delivering supplemental oxygen through a nasal cannula or face mask (target SpO2 >94%).5,8 In addition to relieving symptoms, supplemental oxygen reduces precapillary pulmonary vasoconstriction and RV afterload. Conversely, positive pressure noninvasive ventilation should be used with caution to avoid negative effects on the RV. Pulmonary and systemic congestion should be treated with loop diuretics (and/or vasodilators in selected cases presenting with elevated systolic blood pressure).3,41
Third, anticoagulation should be started as soon as the diagnosis of PE is made or suspected in case of intermediate or high probability (ESC guidelines class I indication).6 In selected patients, oral anticoagulation with direct oral anticoagulants (DOAC) can be used. However, for the majority of patients with PE and AHF, we prefer to start with unfractionated heparin (bolus followed by continuous infusion) then switch to DOAC or vitamin K antagonists (VKA) after a few days. Indeed, in patients with AHF, the bioavailability of oral drugs may be reduced because of gastrointestinal congestion and the renal elimination of DOACs may be impaired if renal function deteriorates because of venous congestion.
The clinical response to the initial treatment should be continuously reevaluated to determine further treatment and patient allocation.42 In case of improvement, diuretics should be further tailored until euvolaemia is achieved; the oral HF therapies should be started or uptitrated and the anticoagulation should be switched to DOAC/VKA.3,37,41
Before hospital discharge, patients should be instructed and a follow-up plan should be organised.
For deteriorating patients, a transfer to the intensive care unit (ICU) or cardiac care unit (CCU) should be considered (see below).
High-risk Patients or With Cardiopulmonary Instability
Patients with either cardiopulmonary instability or with PESI III–V should be managed in the resuscitation area or in the ICU/CCU.5,8,22 In an analogy to the treatment of stable patients, the management of unstable or severely ill patients should consist of three parts delivered simultaneously.
First, the role of additional diagnostic modalities, such as TTE and lung ultrasound, is particularly important in unstable patients to better understand and characterise the underlying cardiac and pulmonary conditions and to optimise cardiopulmonary support. Special attention should be given to signs of RV dysfunction (e.g. RV fractional area change, tricuspid annular plane systolic excursion, tricuspid annular peak systolic velocity and McConnell’s sign), signs of pulmonary and systemic congestion (pulmonary pressure and inferior vena cava diameter), left ventricular systolic and diastolic function, and concomitant valve pathologies. The lung ultrasound may assist differentiation of the cause of hypoxia (pulmonary oedema versus PE).43,44
Second, cardiopulmonary support should include the optimisation of the oxygenation by delivering supplemental oxygen through a nasal cannula or face mask (target SpO2 >94%).5,8 As mentioned above, positive pressure noninvasive/invasive ventilation should be used with caution to avoid negative effects on the RV function. Haemodynamic support should start with optimisation of the preload but we want to add a note of caution here: while some patients presenting with shock may display intravascular fluid depletion and will be fluid responsive, most patients present with systemic congestion and additional fluid loading may worsen organ function.5,21,32
The key step in the management of RV dysfunction is the restoration of the perfusion pressure by adding a vasopressor (i.e. norepinephrine).5,21 Norepinephrine increases the mean arterial pressure, improves coronary perfusion, reduces RV ischaemia without negative effects on RV afterload (pulmonary resistance is not affected by norepinephrine).45 If haemodynamic stability is not restored after optimisation of the preload and vasopressor support and significant RV dysfunction is shown by echocardiography, treatment with a positive inotropic drug to increase myocardial contractility is recommended.5,21 The choice of inotropic drug should be based on the differences in pharmacodynamics and pharmacokinetics.46 Catecholamines (e.g. dobutamine) offer the advantage of a rapid onset of effect (within minutes) but increase myocardial oxygen demand, which can precipitate myocardial ischaemia. Phosphodiesterase III inhibitors (e.g. milrinone) and the calcium-sensitiser levosimendan require longer (hours) to achieve the maximal effect, have a longer duration of effect of hours to days and display strong vasodilatory properties (both substances reduce pulmonary and systemic pressures).47 Levosimendan, in contrast to catecholamines and phosphodiesterase III inhibitors, does not increase myocardial oxygen consumption.
The third part of the treatment of patients with PE and cardiopulmonary instability should be reperfusion strategies. Systemic thrombolysis should be considered in all patients presenting with severe haemodynamic instability or shock caused by PE (ESC guidelines class I indication).6 In this setting, thrombolysis compared to heparin has shown to reduce total mortality, PE-related mortality and PE recurrence, but is associated with a ~10% rate of severe bleedings and a ~2% rate of intracranial haemorrhage.48 It is unclear whether thrombolysis has an impact on symptoms, functional capacity and the development of chronic thrombo-embolic pulmonary hypertension (CTEPH: see below); In our institution, we use alteplase (10 mg as an IV bolus, followed by 90 mg over 2 hours). In patients with a body weight of <65 kg, the maximal dose is 1.5 mg/kg. Of note, unfractionated heparin is not to be withheld during continuous infusion of alteplase.
In high-risk patients who are not in shock or in patients with contraindications for systemic thrombolysis, other options to rapidly reduce the RV afterload might be considered. One option is the use of ultrasound-enhanced, catheter-directed low-dose thrombolysis. However, most knowledge about this technique is derived from registries and case series, and one small randomised trial showing a larger decrease in the RV/LV ratio at 24 hours.49 Surgical pulmonary embolectomy is performed with cardiopulmonary bypass with removal or suction of the fresh clots through incision of the main pulmonary arteries. It provides similar success to systemic thrombolysis and should be considered when systemic thrombolysis is contraindicated (ESC guidelines class I indication).
The re-evaluation of the clinical response to the initial treatment is of crucial importance in unstable patients to timely evaluate the need for mechanical circulatory support, such as extracorporeal life support (ECLS) by the mean of venous–arterial extracorporeal membrane oxygenation (va-ECMO) device.5,21 If clinical improvement is observed, a reduction in cardiopulmonary support should be attempted and, when cardiopulmonary stability is achieved, transfer to a normal ward should be considered.
Long-term Management
The patency restoration of the pulmonary arterial bed occurs within the first weeks to few months in the majority of patients with PE.50,51 However, persistent dyspnoea or poor physical performance months to years after acute PE is frequently reported, particularly in patients with coexisting HF.52 Functional impairment frequently does not correlate with a echocardiography or pulmonary function test, both of which are usually within normal limits.52
In a few PE patients, thrombi become persistent and organised and, in some cases, this may result in the rare but life-threatening condition of CTEPH.6 The hallmark of CTEPH is fibrotic transformation of pulmonary thrombus causing fixed obstruction of some pulmonary arteries and overflow of the other – open – pulmonary arteries, with consecutive microvascular remodelling and the development of pulmonary hypertension. A detailed discussion of diagnosis and treatment of CTPEH is beyond the scope of this article.
We want to emphasise that diagnosing CTEPH is difficult, in particular in patients with other causes of dyspnoea such as HF. Because this condition is rare, we discourage systematic screening with echocardiography in all patients with PE, in particular when asymptomatic. However, in HF patients with recent PE undergoing regular echocardiographic follow-ups, new-onset or worsening of pulmonary hypertension after at least 3 months of anticoagulation therapy may indicate the development of CTPEH and further investigations by right-heart catheter and/or lung scintigraphy are indicated.
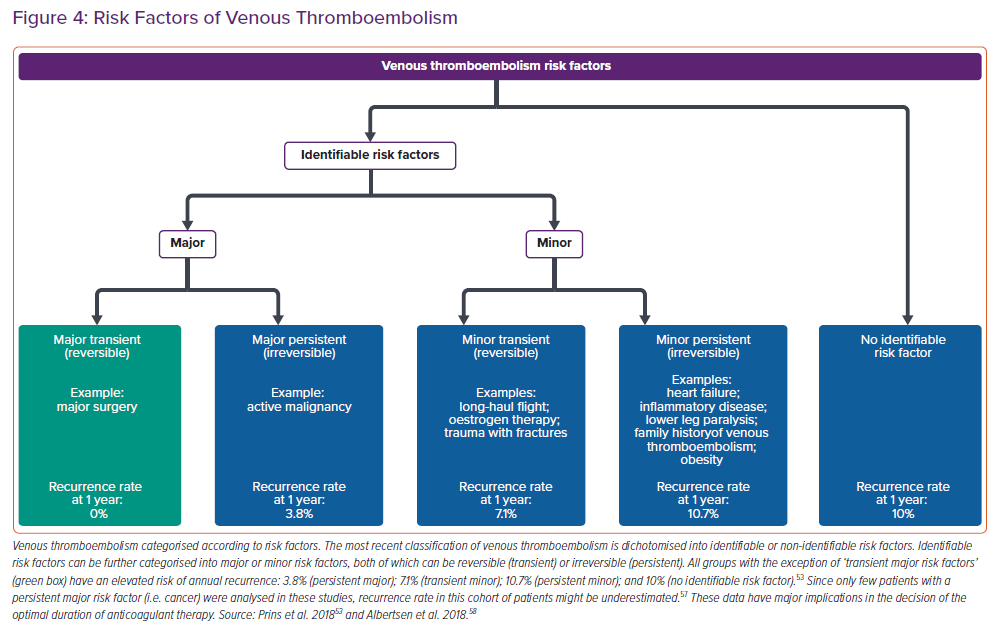
The duration of anticoagulation should be defined after assessment of the risk factors for the development of venous thromboembolism (VTE) (Figure 4). Former classification of VTE using terms as ‘provoked’, ‘unprovoked’ or ‘idiopathic’ VTE should no longer be used since these terms have not proven to be helpful to guide choice and duration of anticoagulation treatment. Of note, except for VTE occurring in the context of a major transient risk factor (e.g. major surgery), in which the risk of recurrence approaches 0% and, as such, can be excluded, all categories have a similar risk of recurrence.53 In other words, the absolute recurrence risk for patients with identifiable risk factors is, in the long term, the same as in patients without identifiable risk factors.
These data support the pathophysiological concept of a final common pathway, by which single risk factors contribute to rather than explain an underlying thrombophilic diathesis. It should further be emphasised that the cumulative incidence of recurrent VTE is continuously increasing over time and a substantial proportion of these recurrent events has fatal outcome resulting in death. In the light of these considerations, anticoagulation for at least 3 months is recommended in all patients with PE (ESC guidelines class I recommendation) and, except for patients with a major transient risk factor, anticoagulation without a predefined date to stop should be the preferred treatment option for patients with VTE in most situations (ESC guidelines class I/IIa recommendation).
In patients with cancer, we prefer to use low-molecular-weight heparin or apixaban for those with gastrointestinal involvement and apixaban or edoxaban for people with non-gastrointestinal cancer.54,55
Conclusion
PE and AHF share many features, including epidemiological aspects, clinical presentation, risk factors and pathobiological mechanisms. Diagnosis and management of these common entities might be challenging for the treating physician, in particular when both conditions are concomitantly present.
The diagnostic approach of HF patients with suspected PE should consider pretest probabilities using clinical gestalt and validated scores. Additional testing (D-dimers and/or CTPA) is employed when needed. Patients with PE and HF should be triaged according to haemodynamic stability. Management should include cardiopulmonary support with special attention to the detrimental effect of PE on RV function, and antithrombotic treatment. Further studies designed to define the best diagnostic and therapeutic approach in this distinct population of patients are needed.